Kristen Gray 0000-0002-1962-189X
Department of Molecular Biology and Biochemistry, Simon Fraser University
Wing Yin Venus Lau 0000-0003-3884-4009
Department of Molecular Biology and Biochemistry, Simon Fraser University
Robert G. Beiko 0000-0002-5065-4980
Faculty of Computer Science, Dalhousie University
Fiona S.L. Brinkman 0000-0002-0584-4099
Department of Molecular Biology and Biochemistry, Simon Fraser University
Abstract
Metagenomic methods enable the simultaneous characterisation of microbial communities without time-consuming and bias-inducing culturing.
Metagenome-assembled genome (MAG) binning methods aim to reassemble individual genomes from this data.
However, the recovery of mobile genetic elements (MGEs), such as plasmids and genomic islands (GIs), by binning has not been well characterised.
Given the association of antimicrobial resistance (AMR) genes and virulence factor (VF) genes with MGEs, studying their transmission is a public health priority.
The variable copy number and sequence composition of MGEs makes them potentially problematic for MAG binning methods.
To systematically investigate this issue, we simulated a low-complexity metagenome comprising 30 GI-rich and plasmid-containing bacterial genomes.
MAGs were then recovered using 12 current prediction pipelines and evaluated.
While 82-94% of chromosomes could be correctly recovered and binned, only 38-44% of GIs and 1-29% of plasmid sequences were found.
Strikingly, no plasmid-borne VF nor AMR genes were recovered, and only 0-45% of AMR or VF genes within GIs.
We conclude that short-read MAG approaches without further optimisation are largely ineffective for the analysis of mobile genes, including those of public-health importance like AMR and VF genes.
We propose that researchers should explore developing methods that optimise for this issue and consider also using unassembled short reads and/or long-read approaches to more fully characterise metagenomic data.
Keywords
Metagenomics, metagenome-assembled genomes, Antimicrobial resistance, Mobile genetic elements, Genomic islands.
Author Notes
* authors contributed equally.
All supporting data, code and protocols have been provided within the article or through supplementary data files. Four supplementary figures and three supplementary tables are available with the online version of this article.
Metagenome assembled genome (MAG) binning has become an increasingly common approach in environmental, microbiome, and public health studies that make use of short-read metagenomic data.
By examining 12 widely-used MAG binning workflows, we demonstrate that these methods are not suitable for the analysis of mobile genetic elements.
Given the potential human and animal health implications of antimicrobial resistance and virulence genes associated with these elements, inappropriate use of short-read MAGs has the potential to be misleading at best and harmful at worst.
This result will hopefully stimulate a fundamental shift in MAG methods to focus on developing methods optimised for these elements as well as incorporating additional read-based and long-read analyses.
Data Summary
In keeping with FAIR principles (Findable, Accessible, Interoperable, Reusable data), all analyses presented in this paper can be reproduced and inspected with the associated github repository https://github.com/fmaguire/MAG_gi_plasmid_analysis (10.5281/zenodo.4005062) and data repository https://osf.io/nrejs/ (10.17605/OSF.IO/NREJS)
Introduction
Metagenomics, the sequencing of DNA from within an environmental sample, is widely used to characterise the functional potential and identity of microbial communities [1, 2].
These approaches have been instrumental in developing our understanding of the distribution and evolutionary history of AMR genes [3–5], as well as tracking pathogen outbreaks [6].
Although long-read DNA technologies (e.g., Oxford Nanopore Technologies’s (ONT) nanopore sequencing [7] and Pacific Biosciences’ (PacBio) single-molecule real-time sequencing [8] platforms) are now being used for metagenomic sequencing [9, 10], high-throughput sequencing of relatively short reads (150-250bp) in platforms such as the Illumina MiSeq still dominates metagenomics.
These reads can be directly analysed using reference databases and a variety of homology search tools (e.g., [11–14]).
Since these reads are shorter than most genes, however, read-based methods provide very little information about their genomic organisation.
This lack of contextual information is particularly problematic in the study of AMR genes and VFs as the genomic context plays a role in function [15], selective pressures [16], and likelihood of lateral gene transfer (LGT) [17].
Sequence assembly using specialised metagenomic de Bruijn graph assemblers (e.g., metaSPAdes [18], IDBA-UD [19], and megahit [20]) is often used to try to recover information about genomic context [21].
To disentangle the resulting mix of assembled fragments, there has been a move to group these contigs based on the idea that those from the same source genome will have similar relative abundance and sequence composition [22].
These resulting groups or “bins” are known as metagenome-assembled genomes (MAGs).
A range of tools have been released to perform this binning including CONCOCT [23], MetaBAT 2 [24], MaxBin 2 [25], and a tool which combines their predictions: DAS Tool [26].
These MAG binning methods have been used successfully in unveiling previously uncharacterised genomic diversity [27–29], but metagenomic assembly and binning has been shown to involve the loss of some information.
This means as little as 24.2-36.4% of reads [30, 31] and ~23% of genomes [31] are successfully assembled and binned in some metagenomic analyses.
The Critical Assessment of Metagenome Interpretation (CAMI) challenge’s (https://data.cami-challenge.org/) Assessment of Metagenome BinnERs (AMBER) [32] benchmarks different MAG recovery methods in terms of global completeness and bin purity.
Similarly, a recent study has also used the AMBER approach to evaluate 15 different binning methods applied to a common metaSPAdes assembly [33].
However, to the best of our knowledge, there has not been a specific assessment of MAG-based recovery of mobile genetic elements (MGEs) such as genomic islands (GIs) and plasmids, despite their health and research importance.
GIs are clusters of chromosomal genes that are known or predicted to have been acquired through LGT events.
GIs can arise following the integration of MGEs, such as integrons, transposons, integrative and conjugative elements (ICEs) and prophages (integrated phages) [34, 35].
They are of high interest since virulence factors (VFs) are disproportionately associated with mobile sequences [36] as well as certain antimicrobial resistance (AMR) genes [37, 38].
GIs often have differing nucleotide composition compared to the rest of the genome [34], a trait exploited by GI prediction tools such as SIGI-HMM [39], IslandPath-DIMOB [40], and integrative tools like IslandViewer [41].
GIs may also exist as multiple copies within a genome [42] leading to potential assembly difficulties and biases in the calculation of coverage statistics.
Plasmids are circular or linear extrachromosomal self-replicating pieces of DNA with variable copy numbers and repetitive sequences [43, 44].
Similar to GIs, the sequence composition (including G+C content, dinucleotide bias, etc.) of plasmids are often markedly different from the genome with which they are associated [45, 46].
Plasmids are also of high interest as a major source of the lateral dissemination of AMR genes throughout microbial ecosystems [37, 48].
GIs and plasmids have proven particularly difficult to assemble from short-read sequencing data.
Due to the history of their integration at specific insertion sites, GIs are commonly flanked by direct repeats [49, 50].
Repetitive sequences are known to complicate assembly from short reads, with repeats often found at contig break sites [51].
Given that assembly of closely related genomes in a metagenome is already challenging [52], the polymorphic nature of GIs and known presence of flanking repeats would be expected to compound these separate assembly issues.
Repeats also inhibit the assembly of plasmids from short read sequencing data, particularly for longer plasmid sequences [53].
Additionally, the varying sequence composition and relative abundance features mean that GIs and plasmids pose significant challenges in MAG recovery.
As these MGEs are key to the function and spread of pathogenic traits such as AMR and virulence, and with MAG approaches becoming increasingly popular within microbial and public-health research, it is both timely and vital that we assess the impact of metagenome assembly and binning on the recovery of these elements.
Therefore, to address this issue we performed an analysis of GI and plasmid, and associated AMR/VF genes, recovery accuracy across a set of 12 state-of-the-art methods for short-read metagenome assemblies.
We show that short-read MAG-based analyses are not suitable for the study of mobile sequences, including those of public-health importance.
Methods
Metagenome Simulation
Thirty RefSeq genomes were selected using IslandPath-DIMOB [40] GI prediction data collated into the IslandViewer database www.pathogenomics.sfu.ca/islandviewer[41] (Supplemental Table 1).
The selected genomes and associated plasmids (listed in Supplemental Table 2 and deposited at osf.io/nrejs/ under “data/sequences”) were manually selected to satisfy the following criteria: 10 genomes with 1-10 plasmids, 10 genomes with >10% of chromosomal DNA predicted to reside in GIs, and 10 genomes with <1% of chromosomal DNA predicted to reside in GIs.
In accordance with the recommendation in the CAMI challenge [52] the genomes were randomly assigned a relative abundance following a log-normal distribution (μ = 1, σ = 2).
Plasmid copy number estimates could not be accurately found for all organisms. Therefore, plasmids were randomly assigned a copy number regime: low (1-20), medium (20-100), or high (500-1000) at a 2:1:1 rate.
Within each regime, the exact copy number was selected using an appropriately scaled gamma distribution (α = 4, β = 1) truncated to the regime range.
Finally, the effective plasmid relative abundance was determined by multiplying the plasmid copy number with the genome relative abundance.
The full set of randomly assigned relative abundances and copy numbers can be found in Supplemental Table 3.
Sequences were then concatenated into a single FASTA file with the appropriate relative abundance.
MiSeq v3 250 base pair (bp) paired-end reads with a mean fragment length of 1000bp (standard deviation of 50bp) were then simulated using art_illumina (v2016.06.05) [54] resulting in a simulated metagenome of 31,174,411 read pairs.
The selection of relative abundance and metagenome simulation itself was performed using the “data_simluation/simulate_metagenome.py” script.
MAG Recovery
Reads were trimmed using sickle (v1.33) [55] resulting in 25,682,644 surviving read pairs.
The trimmed reads were then assembled using 3 different metagenomic assemblers: metaSPAdes (v3.13.0) [18], IDBA-UD (v1.1.3) [19], and megahit (v1.1.3) [20]).
The resulting assemblies were summarised using metaQUAST (v5.0.2) [56].
The assemblies were then indexed and reads mapped back using Bowtie 2 (v2.3.4.3) [12].
Samtools (v1.9) was used to sort the read mappings, and the read coverage was calculated using the MetaBAT2 accessory script (jgi_summarize_bam_contig_depths).
The three metagenome assemblies were then separately binned using MetaBAT2 (v2.13) [24], and MaxBin 2 (v2.2.6) [25].
MAGs were also recovered using CONCOCT (v0.4.2) [23] following the recommended protocol in the user manual.
Briefly, the supplied CONCOCT accessory scripts were used to cut contigs into 10 kilobase fragments (cut_up_fasta.py) and read coverage calculated for the fragments (CONCOCT_coverage_table.py).
These fragment coverages were then used to bin the 10kb fragments before the clustered fragments were merged (merge_cutup_clustering.py) to create the final CONCOCT MAG bins (extra_fasta_bins.py).
Finally, for each metagenome assembly the predicted bins from these three binners (Maxbin2, MetaBAT 2, and CONCOCT) were combined using the DAS Tool (v1.1.1) meta-binner [26].
This resulted in 12 separate sets of MAGs (one set for each assembler and binner pair).
MAG assessment
Synthetic Read Coverage and Depth
The trimmed synthetic reads were mapped back to each reference replicon using bowtie2 (v2.4.1), and sorted and indexed using samtools (v1.10).
The coverage of each reference replicon is calculated using samtools coverage and the per base sequencing depth calculated using samtools depth.
The average and per base depth are then plotted using R (v.3.4.2).
Chromosomal Coverage
The MAG assessment for chromosomal coverage was performed by creating a BLASTN 2.9.0+ [57] database consisting of all the chromosomes of the input reference genomes.
Each MAG contig was then used as a query against this database and the coverage of the underlying chromosomes tallied by merging the overlapping aligning regions and summing the total length of aligned MAG contigs.
The most represented genome in each MAG was assigned as the “identity” of that MAG for further analyses.
Coverage values of less than 5% were filtered out and the number of different genomes that contigs from a given MAG aligned to were tallied.
Finally, the overall proportion of chromosomes that were not present in any MAG was tallied for each binner and assembler.
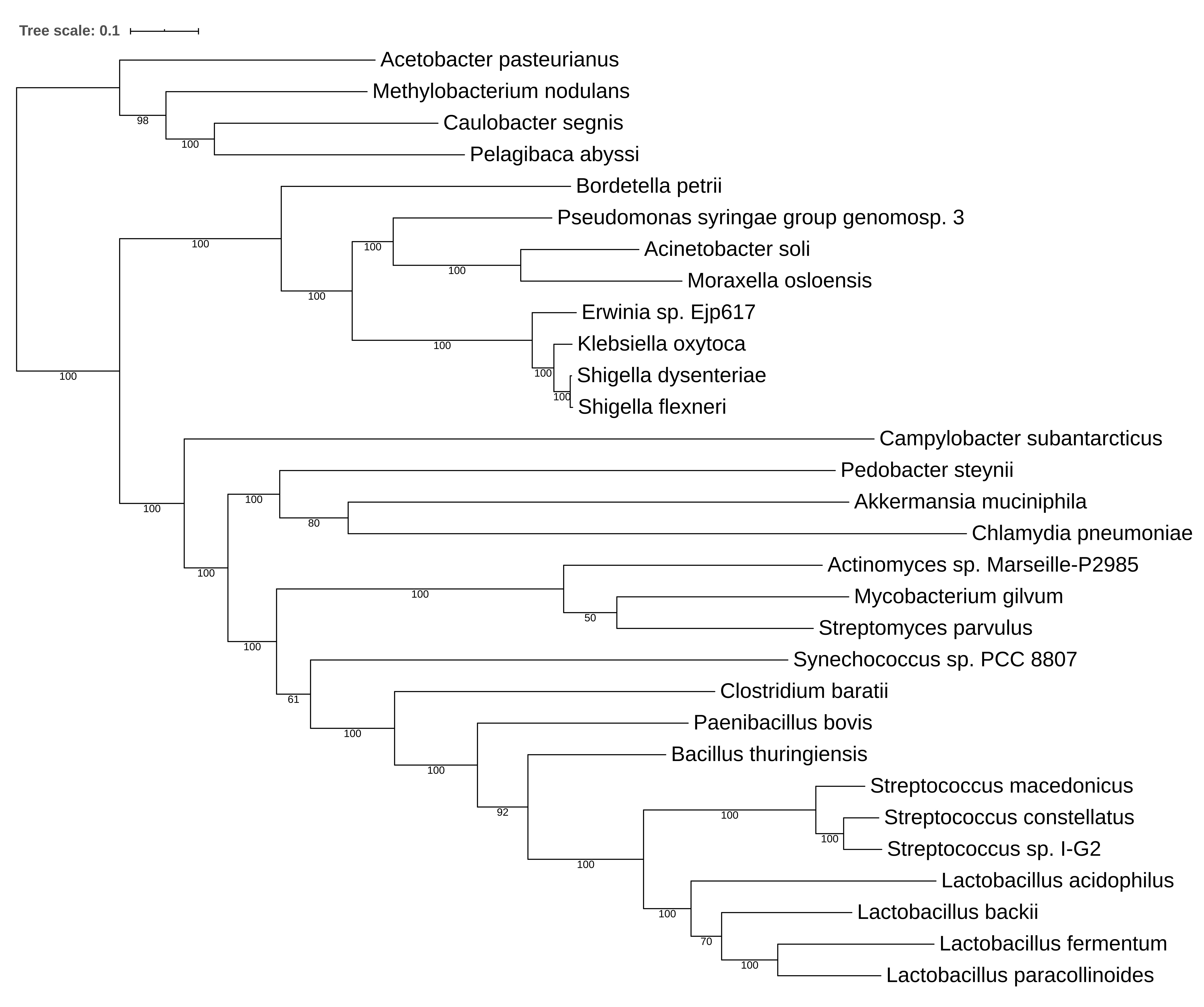
In order to investigate the impact of the presence of closely related genomes in the metagenome on the ability to bin chromosomes we generated a phylogenetic tree for all the input genomes.
Single copy universal bacterial proteins were identified in the reference genomes using BUSCO v4.0.2 with the Bacteria Odb10 data [58].
The 86 of these proteins that were found in every reference genome were concatenated and aligned using MAFFT v7.427 [59] and masked with trimal v1.4.1-3 [60].
A maximum-likelihood phylogeny was then inferred with IQ-Tree v1.6.12 [61] using 1000 ultrafast-bootstraps and the in-built ModelFinder determined partitioning [62].
The phylogeny was then visualised using the interactive Tree of Life (iToL) v4 [63].
Pairwise branch distances were extracted from the resulting tree using ETE3 v3.1.1 [64] and regressed using a linear model against coverage (via seaborn v0.10.0 [65]) and using a Poisson logistic regression model (via statsmodel v0.12.0 [66] against contamination. R^2 and McFadden’s pseudo-R^2 were calculated for each model respectively using the statsmodel library.
Plasmid and GI Coverage
Plasmid and GI coverage were assessed in the same way.
Firstly, a BLASTN database was generated for each set of MAG contigs.
Then each MAG database was searched for plasmid and GI sequences with greater than 50% coverage.
All plasmids or GIs which could be found in the unbinned contigs or MAGs were recorded as having been successfully assembled.
The subset of these that were found in the binned MAGs was then separately tallied.
Finally, we evaluated the proportion of plasmids or GIs that were correctly assigned to the bin that was maximally composed of chromosomes from the same source genome.
Antimicrobial Resistance and Virulence Factors Assessment
Detection of AMR/VF Genes
For the reference genomes, as well as 12 sets of MAGs, prodigal [67] was used to predict open reading frames (ORFs) using the default parameters.
AMR genes were predicted using Resistance Gene Identifier (RGI v5.0.0; default parameters) and the Comprehensive Antibiotic Resistance Database (CARD v3.0.3) [68].
Virulence factors were predicted using the predicted ORFs and BLASTX 2.9.0+ [57] against the Virulence Factor Database (VFDB; obtained on Aug 26, 2019) with an e-value cut-off of 0.001 and a minimum identity of 90% [69].
Each MAG was then assigned to a reference chromosome using the above-mentioned mapping criteria for downstream analysis.
AMR/VF Gene Recovery
For each MAG set, we counted the total number of AMR/VF genes recovered in each metagenomic assembly and each MAG and compared this to the number predicted in their assigned reference chromosome and plasmids.
We then assessed the ability for MAGs to correctly bin AMR/VF genes of chromosomal, plasmid, and GI origin by mapping the location of the reference replicon’s predicted genes to the location of the same genes in the MAGs.
Results
Recovery of Genomic Elements
Chromosomes
The overall ability of MAG methods to recover the original chromosomal source genomes varied widely.
We considered the “identity” of a given MAG bin to be that of the genome that comprises the largest proportion of sequence within that bin.
In other words, if a bin is identifiably 70% species A and 30% species B we consider that to be a bin of species A.
Ideally, we wish to generate a single bin for each source genome consisting of the entire genome and no contigs from other genomes.
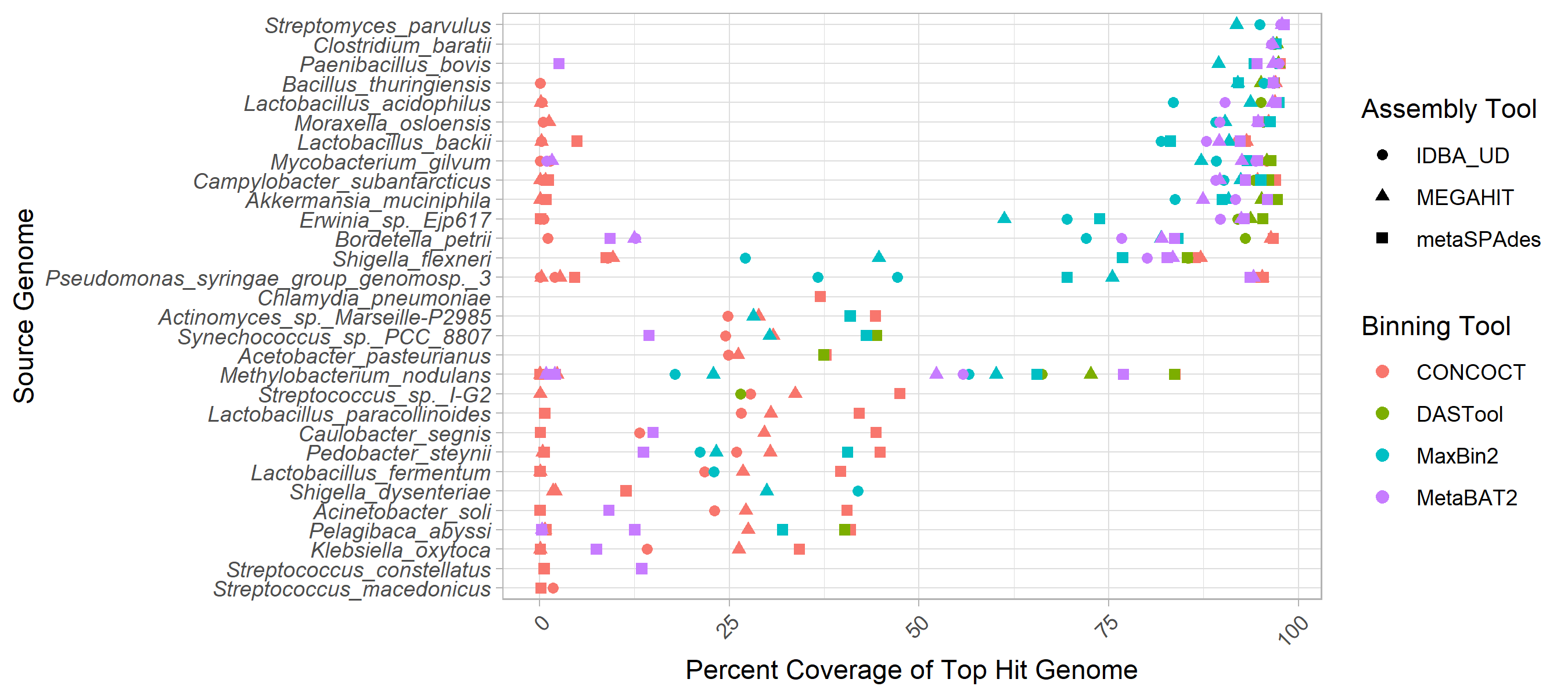
Some genomes are cleanly and accurately binned regardless of the assembler and binning method used (see Fig. 1).
Specifically, greater than 90% of Streptomyces parvulus (minimum 91.8%) and Clostridium baratii (minimum 96.4%) chromosomes are represented in individual bins across all methods.
However, no other genomes were consistently recovered at >30% chromosomal coverage across methods.
The three Streptococcus genomes were particularly problematic with the best recovery for each ranging from 1.7% to 47.49%.
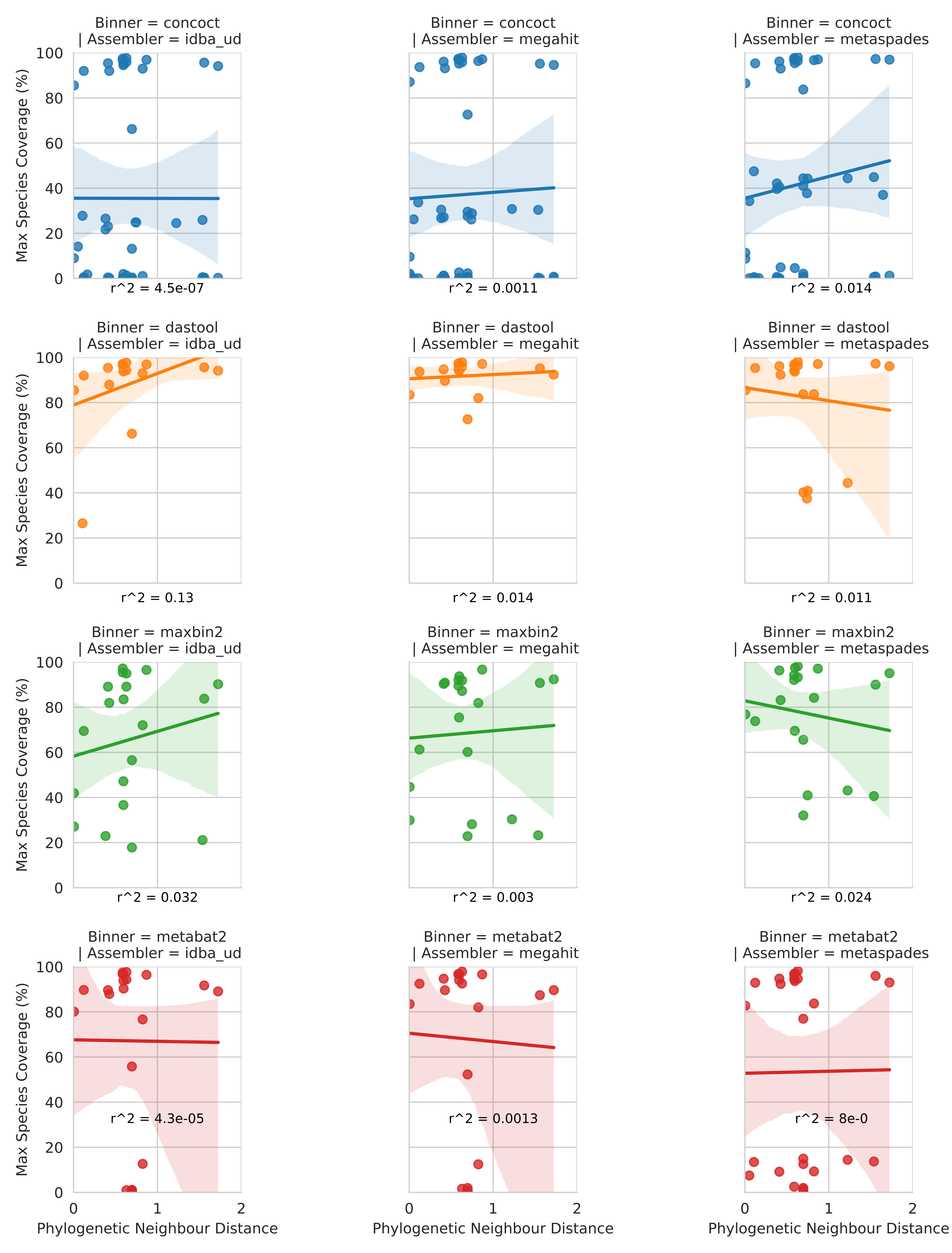
Contrary to what might be expected, the number of close relatives to a given genome in the metagenome did not clearly affect the MAG coverage (Fig. S2).
Figure 1: Top genome coverage for input genomes across MAG binners. Each dot represents the coverage of a specified genome when it comprised the plurality of the sequences in a bin. If a genome did not form the plurality of any bin for a specific binner-assembler pair no dot was plotted for that genome and binner-assembler. The binning tool is indicated by the colour of the dot as per the legend. Genomes such as Clostridium baratti were accurately recovered across all binner-assembler combinations whereas genomes such as Streptococcus macedonicus were systematically poorly recovered.
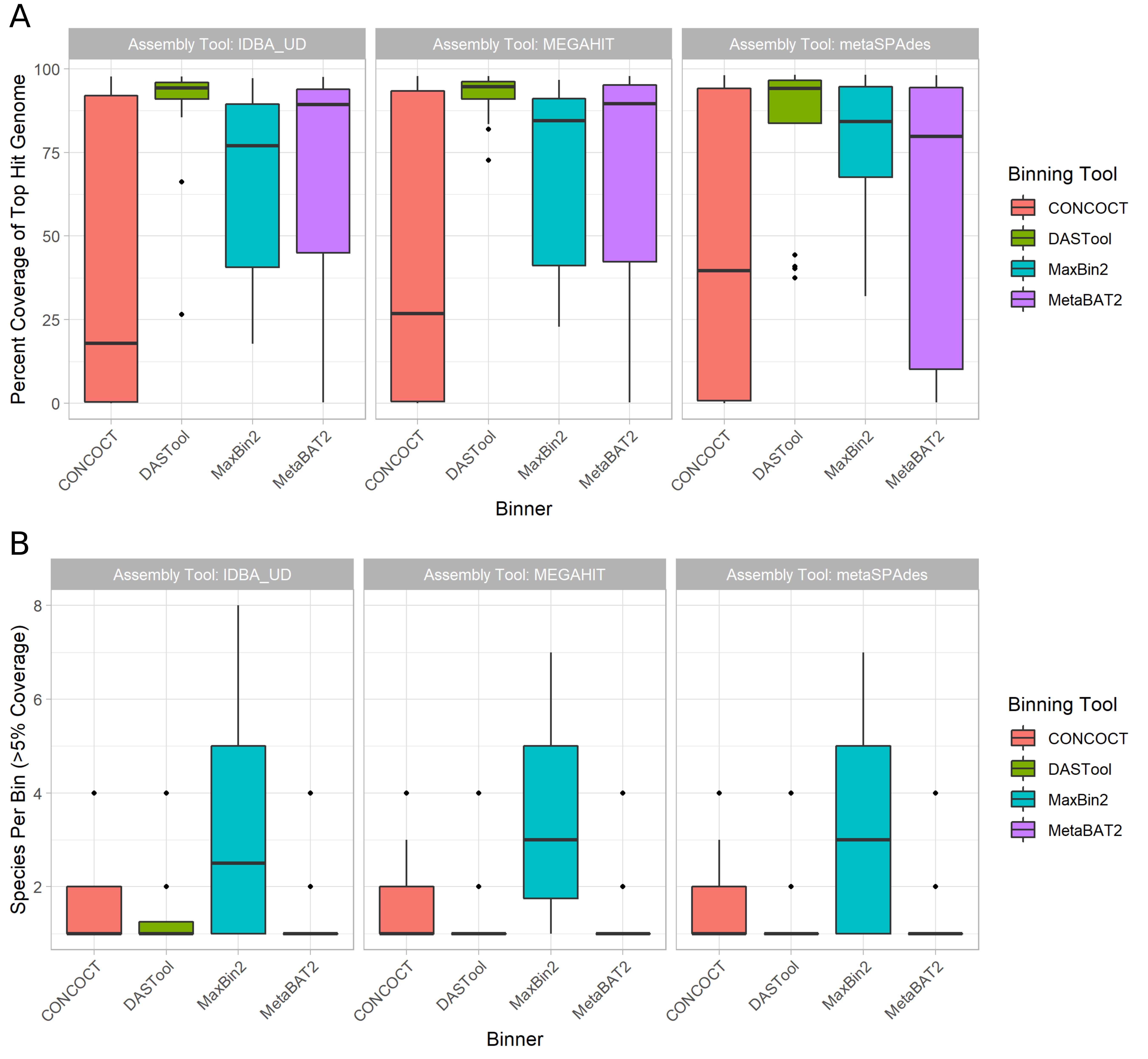
In terms of the impact of different metagenome assemblers, megahit resulted in the highest median chromosomal coverage across all binners (81.9%) with metaSPAdes performing worst (76.8%) (Fig. 2 A).
Looking at binning tools, CONCOCT performed very poorly with a median 26% coverage for top hit per bin, followed by maxbin2 (83.1%), and MetaBAT2 (88.5%).
It is perhaps unsurprising that the best-performing binner in terms of bin top hit coverage was the metabinner DASTool that combines predictions from the other 3 binners (94.3% median top hit chromosome coverage per bin; (Fig. 2 A)).
Figure 2: Overall binning performance for every combination of metagenome assembler (as indicated by pane titles) and MAG binning tool (x-axis and legend colours). Diamonds in the plots represent outliers (greater or lower than the interquartile range marked by the error bars) and the boxes represent the lower quartile, median, and upper quartile respectively. (A) Chromosomal coverage of the most prevalent genome in each bin across binners and metagenome assemblies. Of the 3 assemblers, megahit resulted in the highest median chromosomal coverage (y-axis) across all binners (colored bars) at 81.9% with metaSPAdes performing the worst (76.8%). Of the 4 binners, CONCOCT (red) performed poorly with a median coverage, followed by maxbin2 (blue), MetaBAT2 (purple) and DASTool (green) performing the best.
(B) Distribution of bin purity across assemblers and binners. The total number of genomes present in a bin at >5% coverage (y-axis) was largely equivalent across assemblers (x-axis). For the binning tools, maxbin2 (blue) produced nearly twice as many bins containing multiple species compared to CONCOCT (red), MetaBAT2 (purple) and DASTool (green), which all produced chimeric bins at roughly the same rate.
Bin purity, i.e. the number of genomes present in a bin at >5% coverage, was largely equivalent across assemblers, with a very marginally higher purity for IDBA.
Across binning tools maxbin2 proved an exception with nearly twice as many bins containing multiple species as the next binner (Fig. 2 B).
The remaining binning tools were largely equivalent, producing chimeric bins at approximately the same rates.
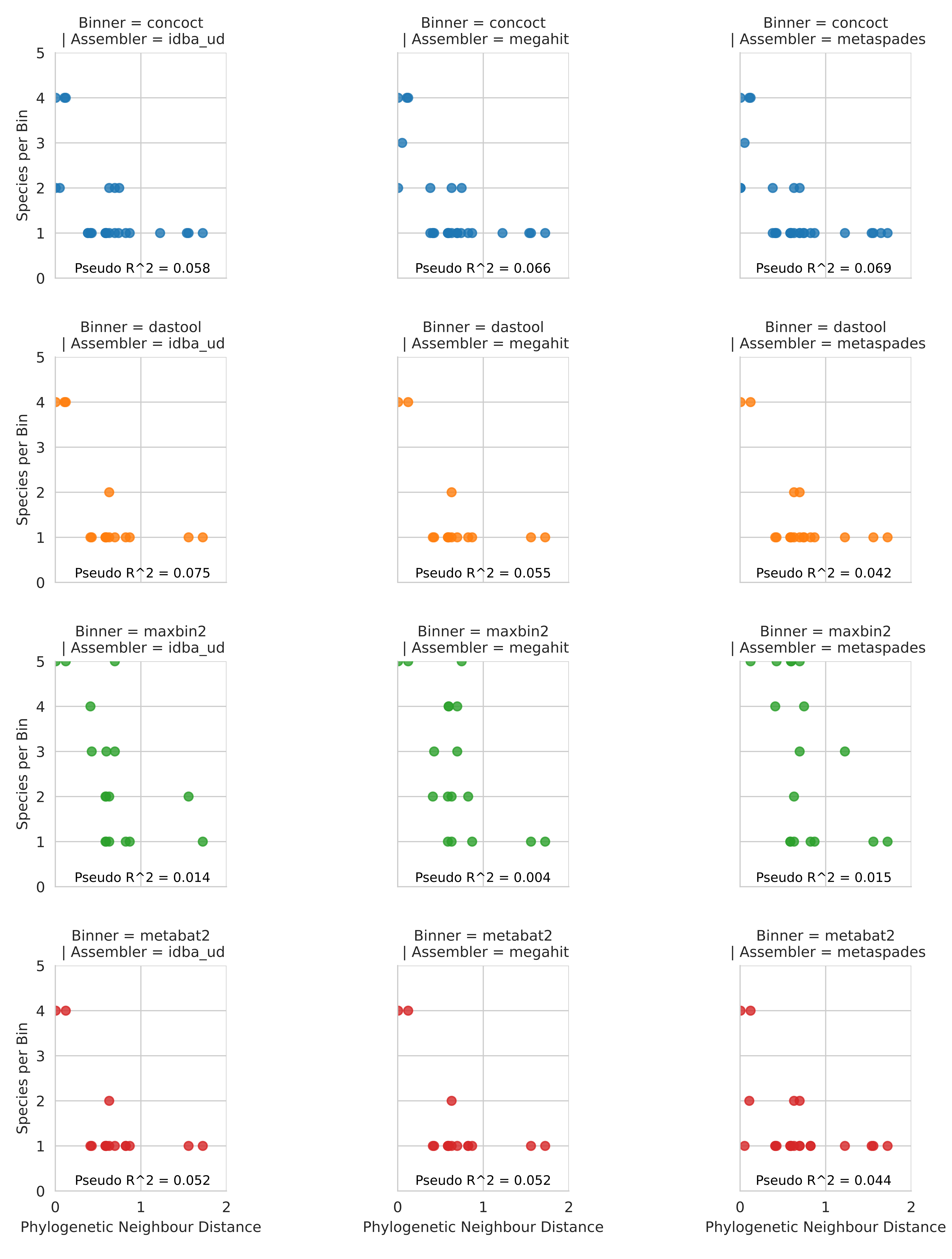
Unlike coverage, purity was potentially affected by the number of close relatives in the metagenome to a given input genome.
Specifically, the closer the nearest relative the less pure the bin (Fig. S3), however, the proportion of variance explained by the regressions were very low for both analyses.
There was also not a clear relationship between coverage of a bin and purity, with low purity but high coverage bins observed, as well as high purity but low coverage bins.
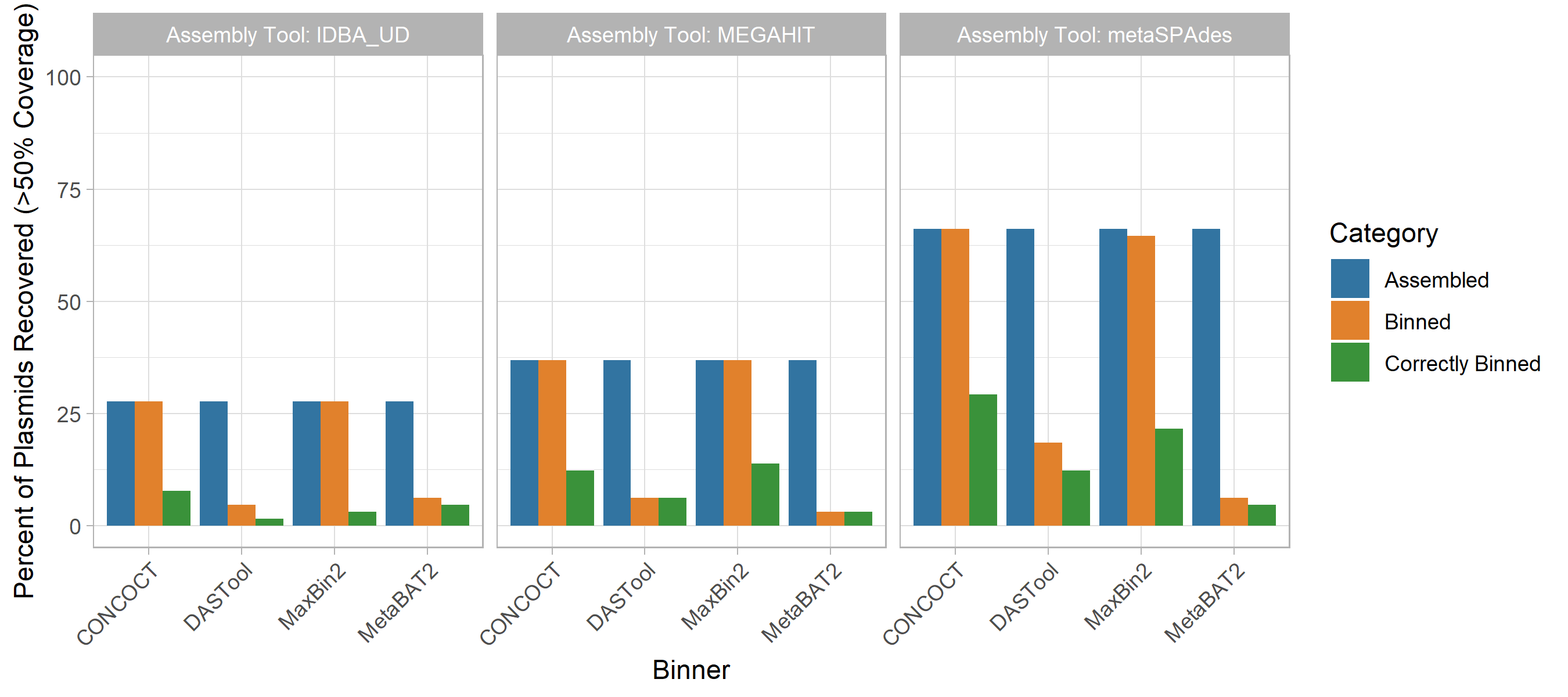
Plasmids
Regardless of method, a very small proportion of plasmids were correctly grouped in the bin that was principally composed of chromosomal contigs from the same source genome.
Specifically, between 1.5% (IDBA-UD assembly with DASTool bins) and 29.2% (metaSPAdes with CONCOCT bins) were correctly binned at over 50% coverage.
In terms of metagenome assembly, metaSPAdes was by far the most successful assembler at assembling plasmids with 66.2% of plasmids identifiable at greater than 50% coverage.
IDBA-UD performed worst with 17.1% of plasmids recovered, and megahit recovered 36.9%.
If the plasmid was successfully assembled, it was, with one exception, placed in a MAG bin by maxbin2 and CONCOCT, although a much smaller fraction were correctly binned (typically less than 1/3rd).
Interestingly, the MetaBAT2 and DASTool binners were more conservative in assigning plasmid contigs to bins; of those assigned to bins, nearly all were correctly binned (Fig. 3).
Figure 3: The performance of metagenomic assembly and binning to recover plasmid sequences. Each plot represents a different metagenome assembler, with the groups of bars along the x-axes showing the plasmid recovery performance of each binning tool when applied to the assemblies produced by that tool. For each of these 12 assembler-binner-pair-produced MAGs the grouped bars from left to right show the percentage of plasmids assembled, assigned to any bin, and binned with the correct chromosomes. These stages of the evaluation are indicated by the bar colours as per the legend. Across all tools the assembly process resulted in the largest loss of plasmid sequences and only a small proportion of the assembled plasmids were correctly binned.
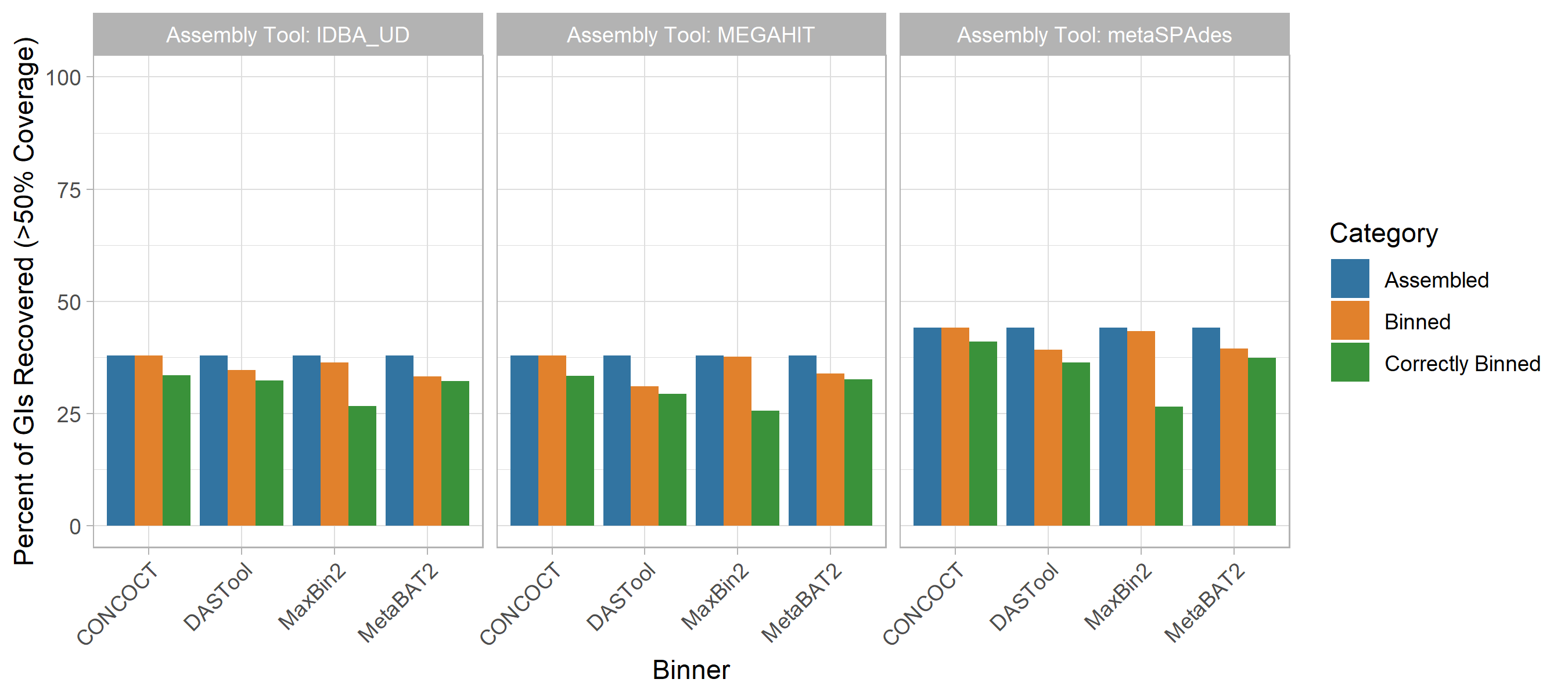
Genomic Islands
GIs were poorly assembled and binned across methods (Fig. 4).
Unlike for plasmids, the performance of different methods was generally less variable, with no clear best-performing method.
Assembly of GIs with >50% coverage was consistently poor (37.8-44.1%) with metaSPAdes outperforming the other two assembly approaches.
For the CONCOCT and maxbin2 binning tools, all GIs that were assembled were assigned to a bin, although the proportion of binned GIs that were correctly binned was lower than for DASTool and MetaBAT2.
DASTool, MetaBAT2 and CONCOCT did not display the same precipitous drop between those assembled and those correctly binned as was observed for plasmids.
In terms of overall correct binning with the chromosomes from the same genome the metaSPAdes assembly with CONCOCT (44.1%) and maxbin2 (43.3%) binners performed best.
Figure 4: Impact of metagenomic assembly and MAG binning on recovery of GIs. GIs were recovered in a similarly poor fashion to plasmids. Generally, <40% were correctly assigned to the same bin majorly comprised of chromosomal contigs from the same source genome regardless of binning (x-axis) and assembly (panel) methods at >50% coverage. metaSPAdes performed the best at assembling GIs (blue). Maxbin2 and CONCOCT placed GIs in a bin majority of the time (orange) however a very small fraction was correctly binned (green). Generally, GIs were correctly binned better than plasmids with DASTool, MetaBAT2 and CONCOCT.
AMR Genes
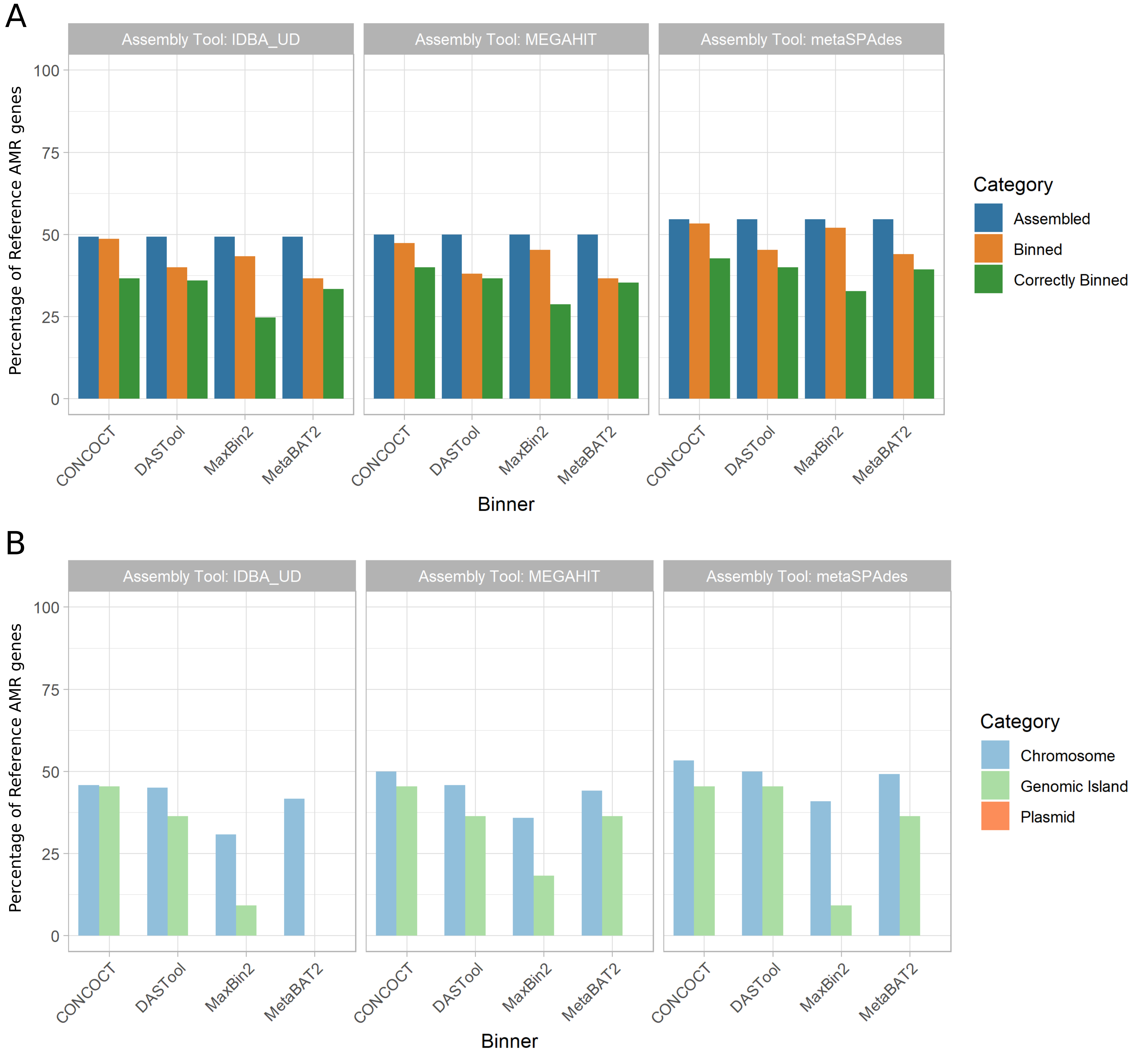
The recovery of AMR genes in MAGs was poor with only ~49-55% of all AMR genes predicted in our reference genomes regardless of the assembly tool used, and metaSPAdes performing marginally better than other assemblers (Fig. 5 A).
Binning the contigs resulted in a ~1-15% loss in AMR gene recovery with the CONCOCT-metaSPAdes pair performing best at only 1% loss and DASTool-megahit performing the worst at 15% reduction of AMR genes recovered.
Overall, only 24% - 40% of all AMR genes were correctly binned.
This was lowest with the maxbin2-IDBA-UD pair (24%) and highest in the CONCOCT-metaSPAdes pipeline (40%).
Figure 5: Recovery of AMR genes across assemblers, binners, and genomic context. (A) The proportion of reference AMR genes recovered (y-axis) was largely similar across assembly tools (panels as indicated by title) at roughly 50% with metaSPAdes performing marginally better overall. Binning tools (x-axis) resulted in a small reduction in AMR genes recovered (orange), however only 24-40% of all AMR genes were correctly binned (green). metaSPAdes-CONCOCT was the best performing MAG binning pipeline. (B) Percent of correctly binned AMR genes recovered by genomic context. MAG methods were best at recovering chromosomally located AMR genes (light blue) regardless of metagenomic assembler or binning tool used. Recovery of AMR genes in GIs showed a bigger variation between tools (light green). None of the 12 evaluated MAG recovery methods were able to recover plasmid located AMR genes.
Moreover, focusing on only the AMR genes that were correctly binned (Fig. 5 B) we can evaluate the impact of different genomic contexts (i.e. chromosomal, plasmid, GI).
Across all methods only 30%-53% of all chromosomally located AMR genes (n=120), 0-45% of GI located AMR genes (n=11) and none of the plasmid-localised AMR genes (n=20) were correctly binned.
Virulence Factor Genes
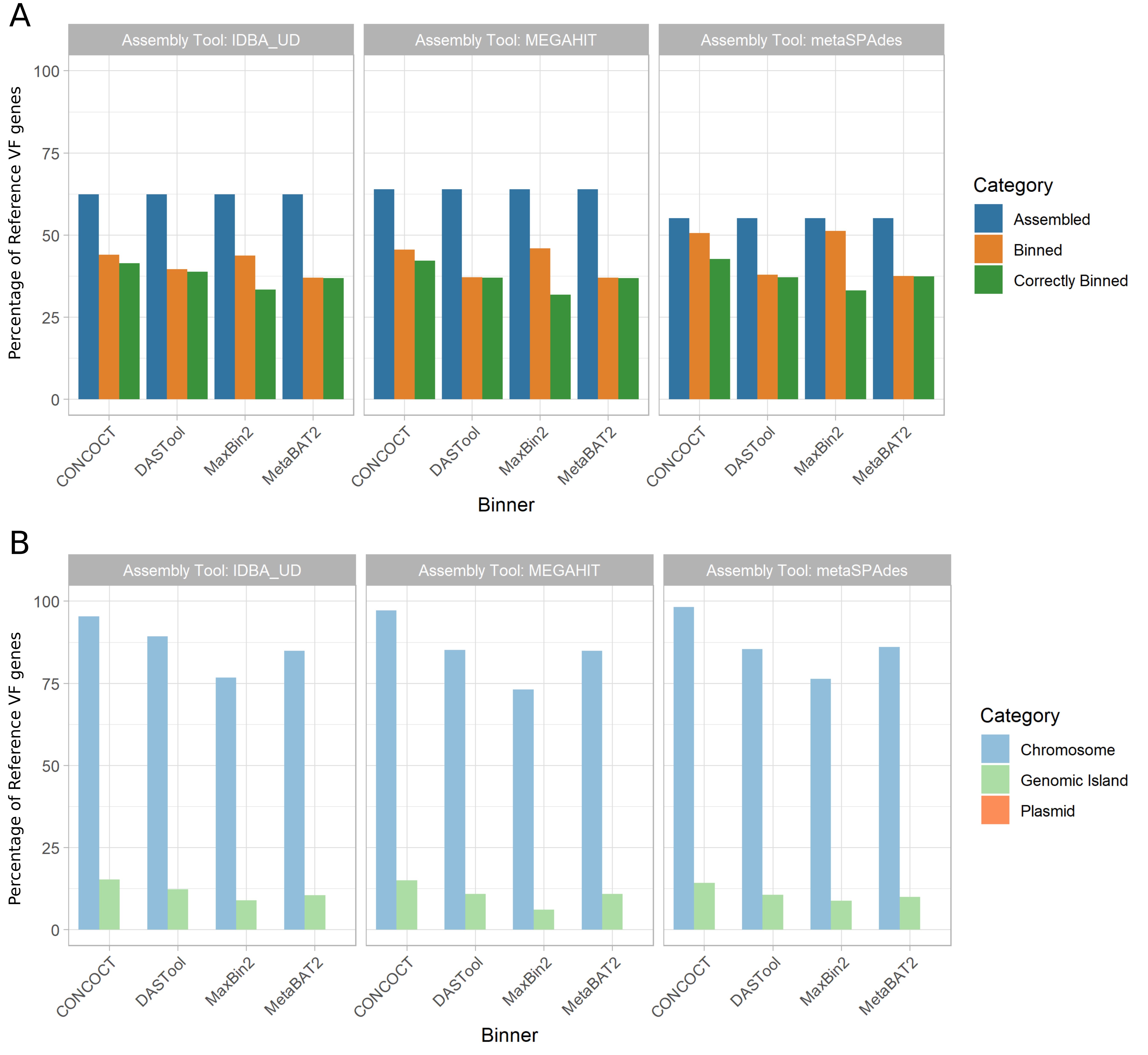
We also examined the impact of MAG approaches on recovery of virulence factor (VF) genes as identified using the Virulence Factor Database (VFDB).
We saw a similar trend as AMR genes (Fig. 6 A).
Between 56% and 64% of VFs were identifiable in the metagenomic assemblies (with megahit recovering the greatest proportion).
The binning process further reduced the number of recovered VFs by 4-26% with DASTool-megahit performing the worst (26% reduction) and CONCOCT-metaSPAdes performing the best (4% reduction).
Unlike AMR genes, the majority of VF genes assigned to a bin were assigned to the correct bin (i.e. that bin largely made up of contigs from the same input genome).
Overall, CONCOCT-metaSPAdes again performed best with 43% of all VFs correctly assigned.
Figure 6: Recovery of VF genes across assemblers, binners, and genomic context. (A) Percent of reference virulence factor (VF) genes recovered across assemblers and binners. The proportion of reference VF genes recovered (y-axis) exhibited a similar trend as AMR genes. Recovery was greatest after the assembling stage (blue), with megahit performing best. Binning tools resulted in a larger reduction in VF genes recovered (orange) compared to AMR genes. However, in the majority of cases, VF genes that are binned are correctly binned (green). metaSPAdes-CONCOCT was again the best performing pair. (B) Percent of correctly binned VF genes recovered in each genomic region. Metagenome assembled genomes (MAGs) were again best at recovering chromosomally located VF genes (light blue), able to correctly bin majority of chromosomally located VFs. GIs recovered again performed very poorly (light green) and again none of the plasmid located AMR genes (orange) was correctly binned.
As with AMR genes, the genomic context (chromosome, plasmid, GI) of a given VF largely determined how well it was binned (Fig. 6 B).
The majority (73%-98%) of all chromosomally located VF genes (n=757) were correctly binned.
However, 0-16% of GI-localised VF genes (n=809) and again none of the plasmid-associated VF genes (n=3) were recovered across all 12 MAG pipelines.
Comparisons of Rates of Loss
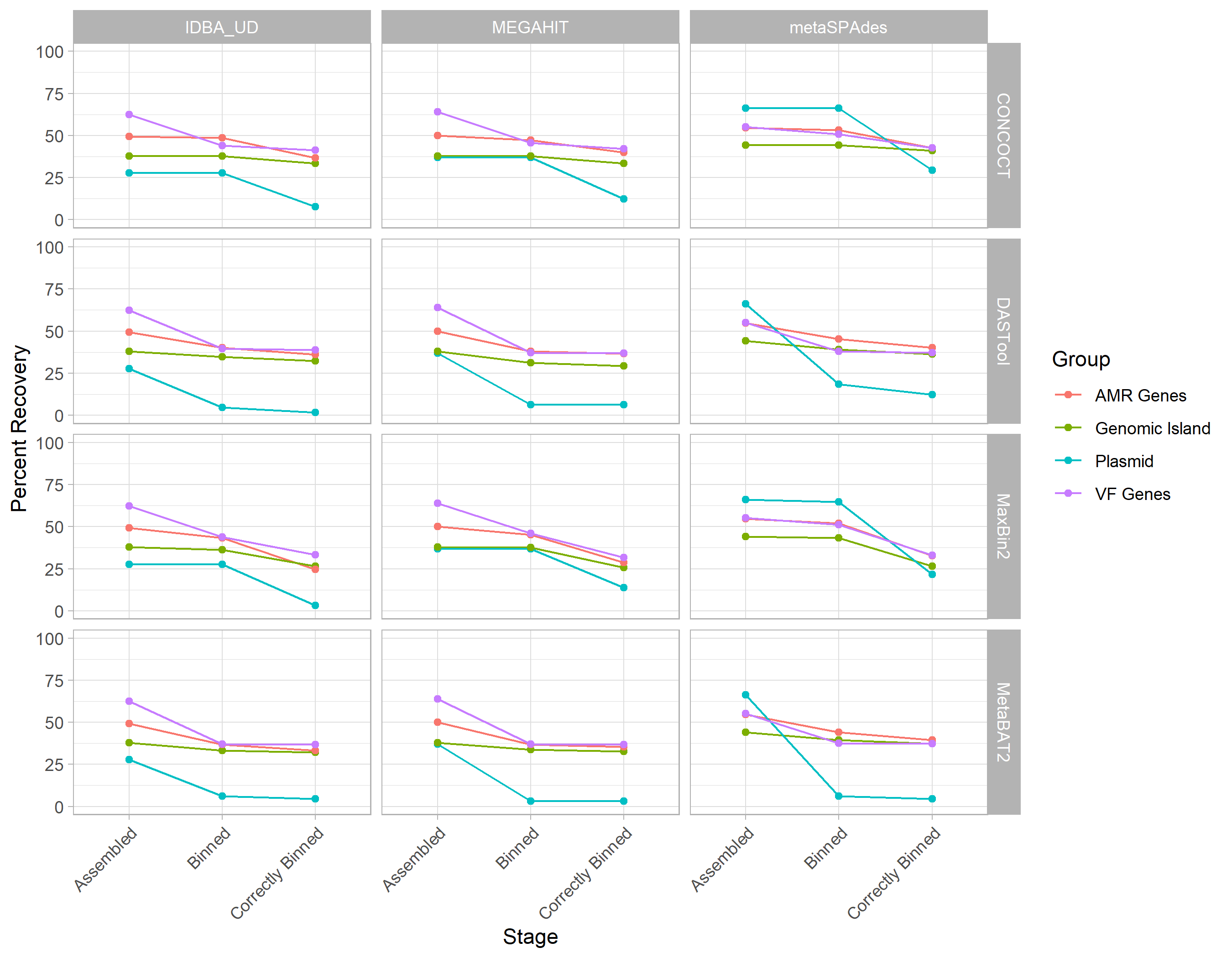
We combined the performance metrics for Figs. 3, 4, 5, and 6 to compare the rates of loss of different components (see Fig. S5).
This highlighted that genomic components (GIs and plasmids) and plasmids in particular are lost at a disproportionately higher rate than individual gene types during MAG recovery.
This also emphasises that better metagenomic assembly doesn’t necessarily result in better binning/recovery of GIs and plasmids.
Simulated Read Analysis
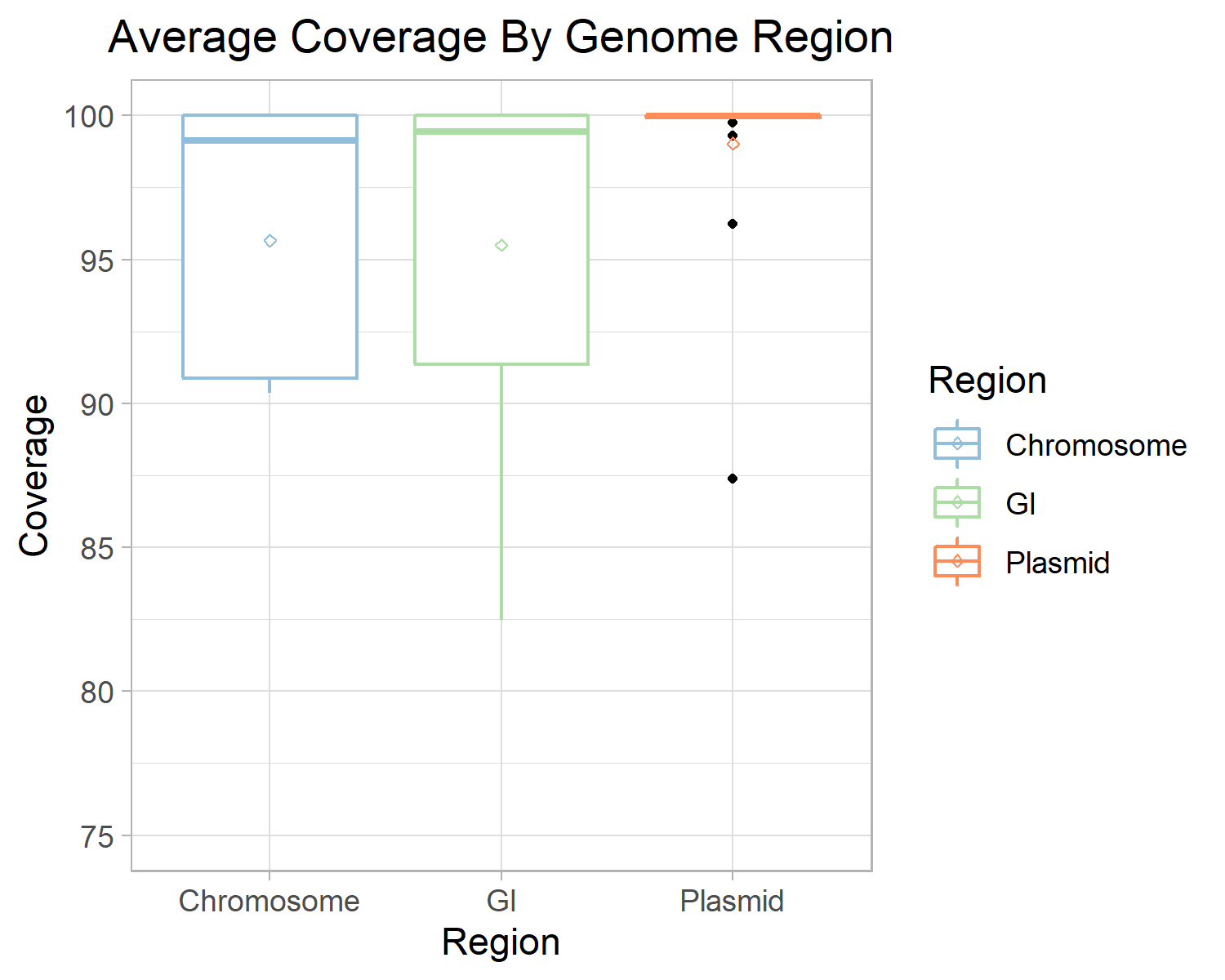
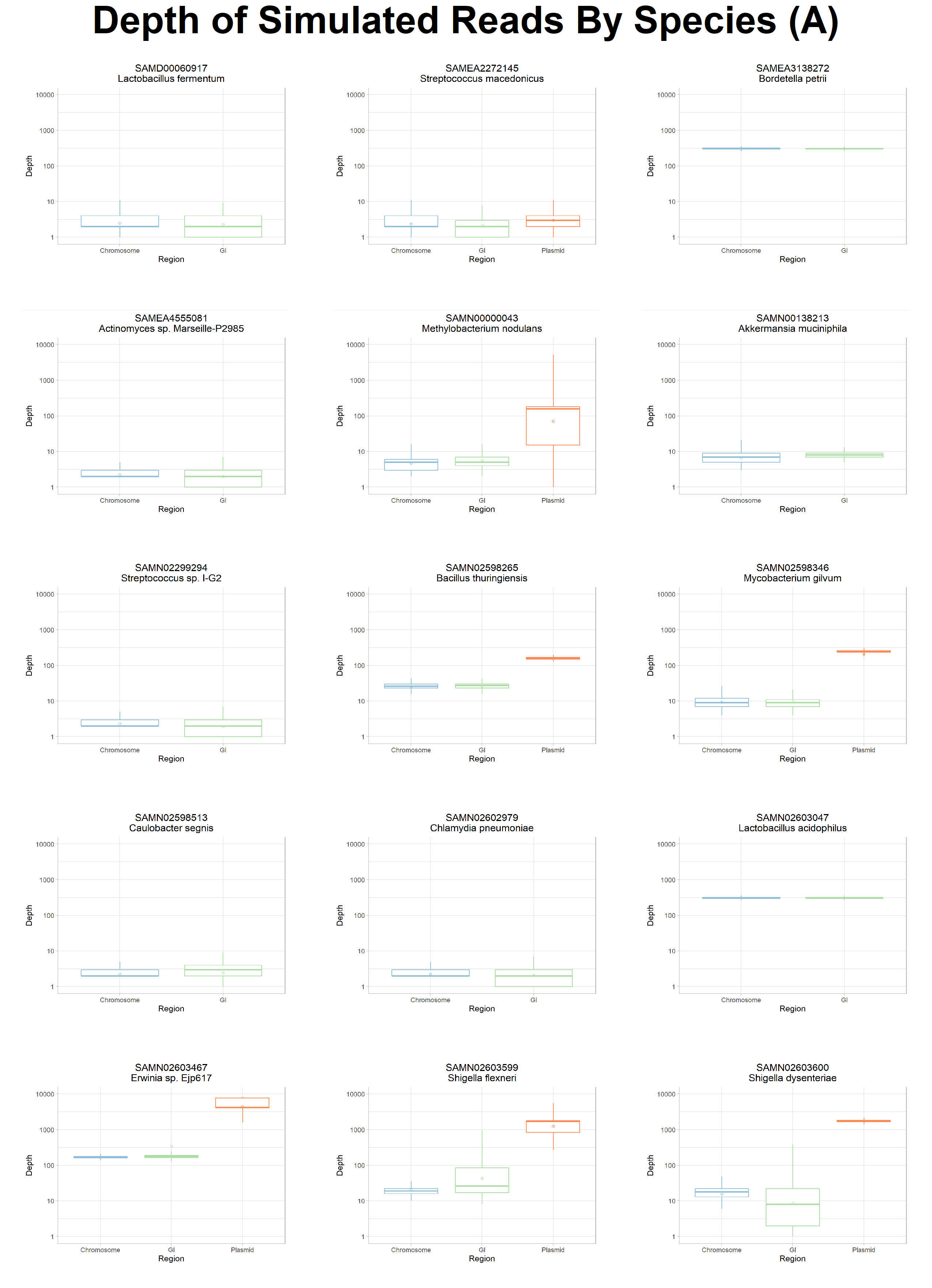
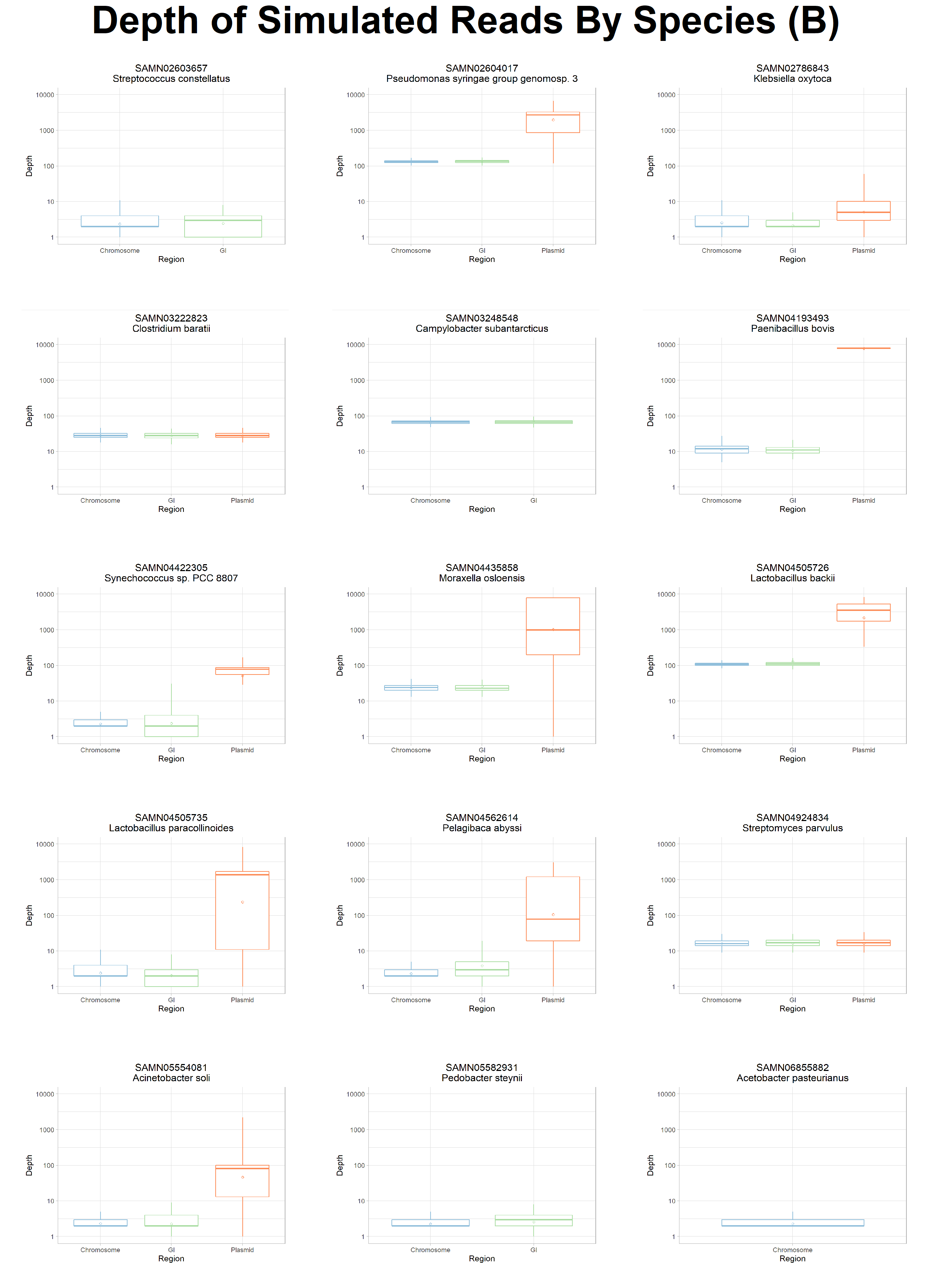
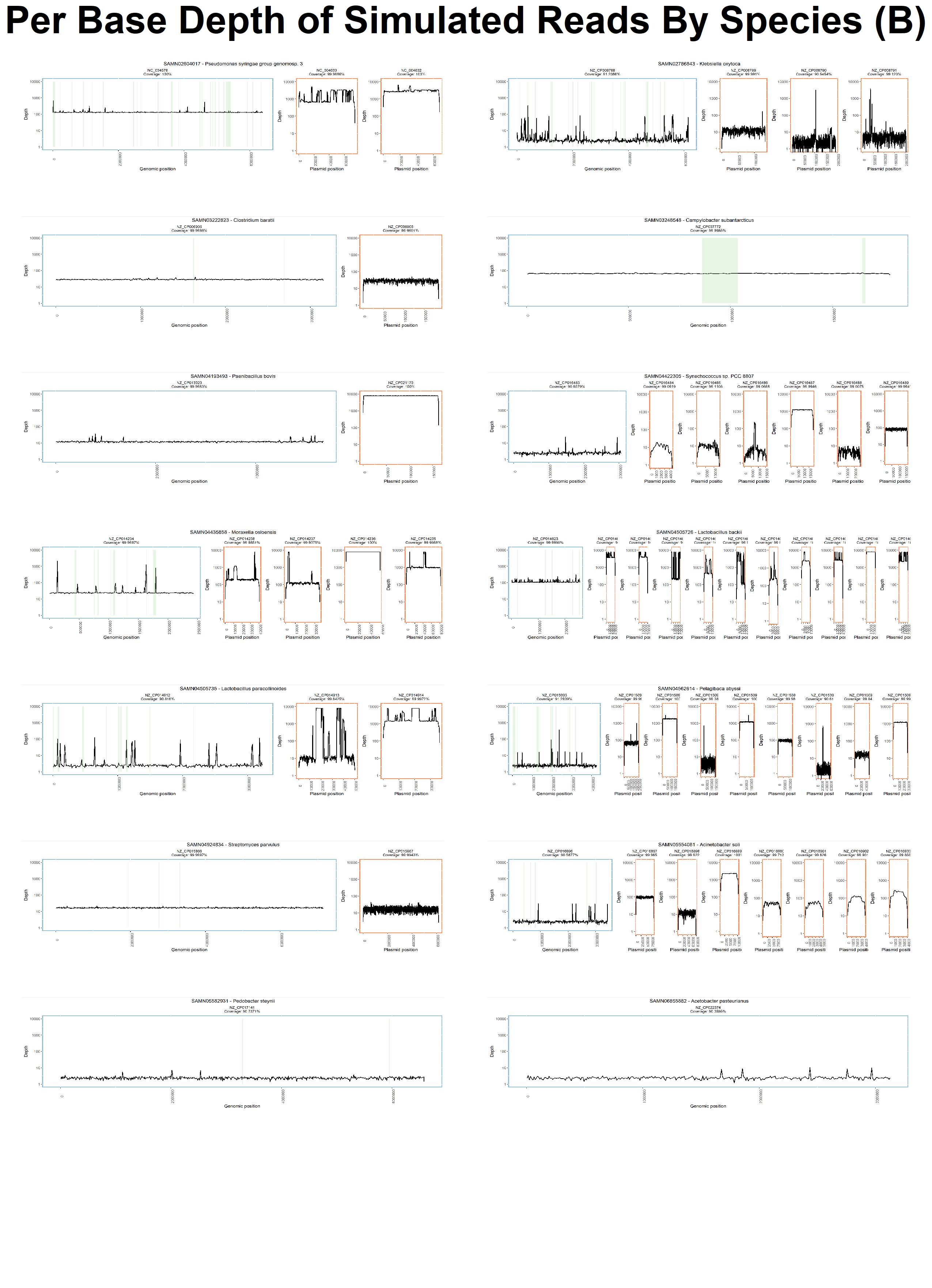
To further explore the potential causes of poor assembly and binning of MGEs we analysed the resultant coverage distribution from mapping our synthetically generated reads back to the original chromosomes, GIs, and plasmids from which they were simulated.
This analysis identified that while coverage of our synthetic metagenomic reads was >96% on average across all reference genomes, the coverage of GIs and plasmids displayed high levels of variance (Fig 7) with large spikes and drops in read depth (see Fig. S7 and S8).
This variability in coverage can be attributed to repeated elements and sequence composition differences that are commonly associated with MGEs.
This issue is likely responsible for failures to accurately estimate the read-depth/coverage in these regions, upon which both assembly (in traversal of the assembly graph) and binning rely.
Figure 7: Average Coverage By Genomic Region. The average coverage of our synthetic reads to their source genome is plotted by their genomic region. Chromosome (blue) and GI (green) displayed a similar average coverage of ~96.5%. Plasmids (orange) had a higher average coverage at ~98%. The per-genome coverage range of plasmids and GIs are higher than chromosomes. Diamond dot indicates the mean coverage of a region and black dots indicate outliers.
Discussion
In this paper, we evaluated the ability of metagenome-assembled genome (MAG) binning methods to correctly recover mobile genetic elements (MGEs; i.e. GIs and plasmids) from metagenomic samples.
Overall, chromosomal sequences were binned well (up to 94.3% coverage, with perfect bin purity using megahit-DASTool).
The presence of closely related genomes had unclear impacts on coverage and cross-contamination of bins (e.g. Streptococcus species in Fig. S2, S3).
Additionally, the trade-off between false positives and sensitivity in the binning of closely related sequences is an area in need of further exploration.
Given the importance of MGEs in the function and spread of virulence traits and AMR, it is particularly noteworthy that regardless of MAG binning method, plasmids and GIs were disproportionately lost compared to core chromosomal regions.
At best (with metaSPAdes and CONCOCT) 29.2% of plasmids and 44.1% of GIs were identifiable at >50% coverage in the correct bin (i.e. grouped with a bin that was mostly made up of contigs from the same genome).
While some MGEs were likely recovered in more partial forms (<50% coverage), use of these by researchers interested in selective pressures and lateral gene transfer could lead to inaccurate inferences.
This poor result is congruent with the intuition that the divergent compositional features and repetitive nature of these MGEs is problematic for MAG methods (a conclusion supported by the observed high coverage variability of MGEs when mapping simulated reads back to the original genomes).
The particularly poor plasmid binning performance is likely attributable to the known difficulties in assembly of plasmids from short-read data [53].
Therefore, binning efficiency might improve with use of long-read sequencing or assembly methods optimised for the assembly and binning of plasmid sequences [53] (such as SCAPP [70]).
Despite its lower effective sequencing depth and higher error rates, incorporating long-read data has been shown to improve overall MAG binning [71] and facilitate metagenomic characterisation of plasmids [72].
However, the low throughput and high error rate of current long-read technologies relative to widely used short-read approaches present a challenge when characterising MGEs in metagenomes, especially those of greater complexity.
Further research is needed to fully characterise the performance of different long-read protocols and analytical approaches (including hybrid approaches with short-reads) on the accuracy of recovering MGEs in metagenomic samples.
With the growing use of MAG methods in infectious disease research (e.g., [73–77]) and the public-health and agri-food importance of the LGT of AMR and VF genes, we also specifically evaluated the binning of these gene classes.
The majority of these genes were correctly assembled across assemblers but were either not assigned or incorrectly assigned to MAG bins during binning.
At best across all binners, 40% of all AMR genes and ~63% of VF genes (CONCOCT-metaSPAdes) present in the reference genomes were assigned to the correct MAG.
While a majority of chromosomally located VF genes (73-98%) and AMR genes (53%) were binned correctly, only 16% of GI VFs (n=809), 45% of GI AMR genes (n=11), and not a single plasmid associated VF (n=3) or AMR gene (n=20) were correctly binned.
This included critical high-threat MGE-associated AMR genes such as oxacillinases (OXA) and Klebsiella pneumoniae carbapenemases (KPC).
One potential caveat of this is that some AMR genes and VFs may no longer be detectable in MAGs due to issues with ORF prediction (see supplementary information & Fig.
S3).
We also observed a higher variability in both the read depth and read coverage in MGEs (Fig. 7, S7, and S8).
This, combined with previous studies observing fragmented ORF predictions in draft genomes, can lead to downstream over- or under-annotation with functional labels depending on the approach used [78].
Although not yet developed, methods that combine the assembly/binning pipelines tested here with read-based inference would provide a better sense of which functions are potentially being missed by the MAG reconstructions.
Our simulated metagenomic community comprised 30 distinct bacterial genomes with varying degrees of relatedness.
While this diversity can be representative of certain clinical samples [79–81], other environments with relevance to public health such as the human gut, soil, and livestock can have 100-1000s of species [82–85].
In addition, MGEs such as GIs and plasmids are known to recombine, producing closely related variants [86–88] that could further complicate assembly from a metagenomic sample.
Polymorphic MGEs were not explicitly introduced in our simulated metagenome.
Consequently, our analysis likely over-represents the effectiveness of the methods tested in a public-health setting.
Metagenomic simulation is also unlikely to perfectly represent the noise and biases in real metagenomic sequencing but it does provide the ground-truth necessary for evaluation [32, 89].
This simulation approach, combined with the development of an MGE/AMR-focused mock metagenome (similarly to the mockrobiota initiative [90]), could provide a key resource to develop and validate new binning approaches and different sequencing strategies.
Additionally, it would provide a way to further optimise parameter settings of existing metagenomic assembly and binning tools beyond the default settings used in these analyses (considered representative of most “real-world” usage [91]) without overfitting to a particular metagenome.
This study has shown that while short-read MAG-binning approaches provide a useful tool to study a bacterial species’ core chromosomal elements they have severe limitations in the recovery of MGEs.
The majority of these MGEs will either fail to be assembled or be incorrectly binned.
The consequence of this is the disproportionate loss of key public-health MGE-associated VFs and AMR genes that may be crucial markers for monitoring the spread of virulence and resistance among clinically important pathogens.
As many of these clinically relevant genes have a high propensity for lateral gene transfer between unrelated bacteria [36, 37] it is critical to highlight that MAG approaches alone are currently insufficient to thoroughly profile them.
Within public-health metagenomic research, as well as other research areas that study MGEs, it is vital we utilise MAGs in conjunction with other methods (e.g. targeted AMR [92], long-read sequencing, plasmid specialised assembly approaches [70], and read-based sequence homology search [11]) before drawing biological or epidemiological conclusions.
Supplementary Information
Impact of Related Genomes on MAG binning
By generating a phylogeny of universal single copy genes in our input genomes (Fig. S1) we analysed the relationship between the presence of closely related genomes and the ability of the different MAG-recovery methods to bin chromosomal sequences.
Specifically, we regressed phylogenetic distance on this phylogeny with per-bin chromosomal coverage (Fig. S2) and bin purity (Fig. S3).
This identified no clear relationship between chromosomal coverage (Fig. S2), or purity (Fig. S3), and the phylogenetic distance to the nearest relative in the metagenome.
Figure S1: Unrooted universal single-copy gene concatenation maximum-likelihood (IQ-TREE) phylogeny. Percentage of ultrafast-bootstraps (n=1000) supporting each bifurcation are annotated on each node. Phylogeny was visualised using iToL.
Figure S2: Relationship between phylogenetic distance to closest neighbour input genome on genomic coverage in MAG majority comprised of that taxon. Each dot represents the genomic coverage of a particular genome and the branch distance on an 86-protein concatenated phylogeny between that genome and its nearest neighbour. Rows indicate the binning software and columns the metagenomic assembler. Regression line is a simple linear model fitted in seaborn with R^2 values calculated and annotated on each plot.
Figure S3: Relationship between phylogenetic distance to closest neighbour input genome on bin purity. Each dot shows the number of other input genomes detectable in a given MAG bin in relation to the branch distance on an 86-protein concatenated phylogeny between the majority genome in that bin and its nearest neighbour. McFadden’s pseudo-R^2 calculated from fitted poisson logistic regression models are annotated on each plot.
Recovery of Specific Gene Content
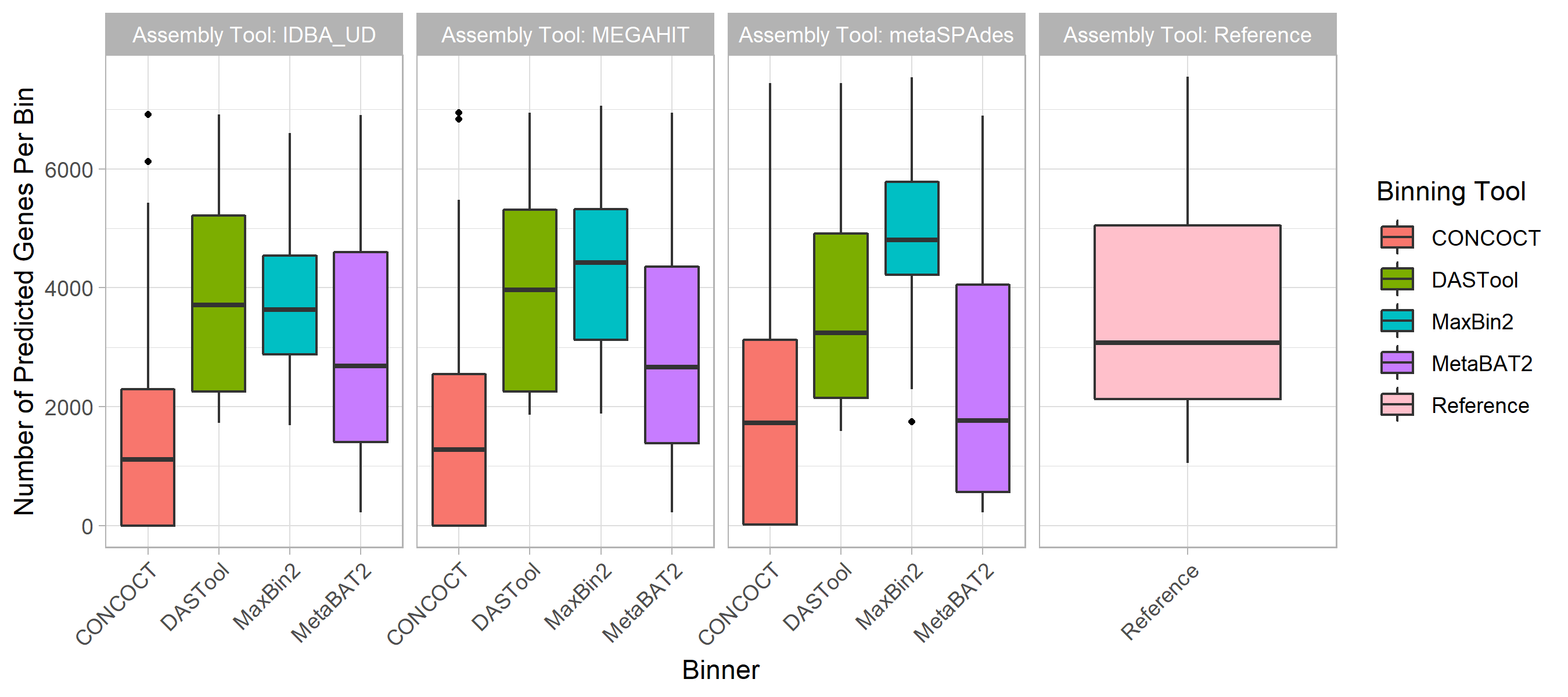
We explored the ability of different approaches to find open reading frames (ORFs) within MAGs.
Overall, the total number of predicted ORFs in MAGs followed a similar trend (Fig. S4) as the chromosomal coverage and purity (Fig. 2).
Of the four binning tools, CONCOCT performed the worst, finding <30% of the number of ORFs in our reference genomes used to construct the synthetic data.
MetaBAT2 performed second worst at ~80%.
DASTool recovered a similar number to our reference and Maxbin2 detected 7-46% more genes.
The Assembler method did not significantly impact the number of genes predicted with the exception of Maxbin2, in which IDBA_UD was the closest to reference and metaSPAdes predicted 46% more ORFs.
Given that there is reason to suspect that there are some issues with the ORF calling in the MAGs. i.e. some tools produced more predicted ORFs than reference, it could be the case that some of these sequences are present in the assemblies (with errors/gaps), but are not being identified as ORFs, or are broken into multiple ORFs, leading to issues downstream labeling them correctly as AMR/VF genes.
Regardless of different tools producing a different number of ORFs, the recovery of AMR/VF is pretty consistent regardless of how many ORFs are predicted.
Figure S4: Predicted Gene Content. The total number of open reading frames (ORF) predicted followed the same trend as chromosomal coverage and purity. The assemblers (colored bars) did not contribute to variability in the number of ORFs detected. Of the 4 binners, CONCOCT recovered <30% of our reference genome ORFs. DASTool and MetaBAT2 predicted a similar number as our reference genomes.
Comparisons of Rates of Loss
Combining the performance metrics for Figs. 3, 4, 5, and 6 to compare the rates of loss of different components emphasises some of the observed patterns (see Fig. S5).
This highlights that genomic components (GIs and plasmids) and plasmids in particular are lost at a higher rate than individual gene types during MAG recovery.
Figure S5: Comparison of rates of loss for different genomic components and gene types across assemblers and binning tools. Each line represents a different component as indicated by the legend with assemblers indicated by row and binning tool by column. This shows that regardless of approach genomic components (GIs and plasmids) are lost at a higher rate than individual VF or AMR genes.
Figure S6: Predicted Gene Content. The total number of open reading frames (ORF) predicted followed the same trend as chromosomal coverage and purity. The assemblers (colored bars) did not contribute to variability in the number of ORFs detected. Of the 4 binners, CONCOCT recovered <30% of our reference genome ORFs. DASTool and MetaBAT2 predicted a similar number as our reference genomes.
Detailed Simulated Read Depth Analysis
Figure S7A:
Figure S7: Average Read Depth Per Genome. Across all of the reference species (facet), the read depth of plasmids (orange) is considerably higher relative to chromosomes (blue), likely due to the copy number regime randomly assigned. GIs (green) exhibited relatively lower read depth compared to chromosomes. The variability in read depth is notably higher in and around GIs and plasmids.
Figure S8A:
Figure S8: Per Base Read Depth Per Species. The per base (x-axis) read depth (y-axis) of each species is plotted individually. Overall, the read read depth of chromosomes (blue boxes) is much lower than read depth of plasmids (orange boxes). GIs within the chromosome are highlighted in green. At a per base level, we see a much lower read depth at the beginning and the end of each replicon as well as a higher variability in read depth for GIs and plasmids.
This work was supported primarily by a Donald Hill Family Fellowship held by F.M.
W.Y.V.L. and B.J. hold Canadian Institutes of Health Research (CIHR) doctoral scholarships.
K.G. was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) Collaborative Research and Training Experience (CREATE) Bioinformatics scholarship.
B.J, W.Y.V.L., and K.G. also held Simon Fraser University (SFU) Omics and Data Sciences fellowships.
F.S.L.B. holds an SFU Distinguished Professorship and R.G.B. is a Professor and Associate Dean Research at Dalhousie University.
Additionally, this work was partially supported by Genome Canada and NSERC grants to R.G.B. and F.S.L.B.
Acknowledgements
The authors would like to thank their funders and the Simon Fraser University (SFU) Research Computing Group and Compute Canada for compute resource support.
Author contributions
F.M. and B.J.: conceptualization, investigation, validation, formal analysis, data curation, writing (original draft preparation; review and editing), visualization.
W.Y.V.L. and K.G.: investigation, data curation writing (review and editing).
F.S.L.B and R.G.B.: Supervision, Project administration, funding, writing (review and editing).
All authors contributed to and approved the manuscript.
Conflict of Interest
The authors declare no competing interests.
References
1. Breitbart M, Salamon P, Andresen B, Mahaffy JM, Segall AM et al. Genomic analysis of uncultured marine viral communities. Proceedings of the National Academy of Sciences 2002;99:14250–14255.
2. Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nature Biotechnology 2017;35:833–844.
3. Donia M, Cimermancic P, Schulze C, Wieland Brown L, Martin J et al. A Systematic Analysis of Biosynthetic Gene Clusters in the Human Microbiome Reveals a Common Family of Antibiotics. Cell 2014;158:1402–1414.
4. D’Costa VM, Griffiths E, Wright GD. Expanding the soil antibiotic resistome: exploring environmental diversity. Current Opinion in Microbiology 2007;10:481–489.
5. D’Costa VM, King CE, Kalan L, Morar M, Sung WWL et al. Antibiotic resistance is ancient. Nature 2011;477:457–461.
6. Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M et al. A Culture-Independent Sequence-Based Metagenomics Approach to the Investigation of an Outbreak of Shiga-Toxigenic Escherichia coli O104:H4. JAMA 2013;309:1502.
7. Mikheyev AS, Tin MMY. A first look at the Oxford Nanopore MinION sequencer. Molecular Ecology Resources 2014;14:1097–1102.
8. Eid J, Fehr A, Gray J, Luong K, Lyle J et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 2009;323:133–138.
9. Nicholls SM, Quick JC, Tang S, Loman NJ. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. GigaScience;8. Epub ahead of print May 2019. DOI: 10.1093/gigascience/giz043.
10. Somerville V, Lutz S, Schmid M, Frei D, Moser A et al. Long-read based de novo assembly of low-complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. BMC Microbiology;19. Epub ahead of print 25 June 2019. DOI: 10.1186/s12866-019-1500-0.
11. Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nature Methods 2014;12:59–60.
12. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods 2012;9:357–359.
13. Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013;29:2487–2489.
14. Ounit R, Wanamaker S, Close TJ, Lonardi S. CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics;16. Epub ahead of print 25 March 2015. DOI: 10.1186/s12864-015-1419-2.
15. Xu X, Lin D, Yan G, Ye X, Wu S et al. vanM, a New Glycopeptide Resistance Gene Cluster Found in Enterococcus faecium. Antimicrobial Agents and Chemotherapy 2010;54:4643–4647.
16. Baker-Austin C, Wright MS, Stepanauskas R, McArthur JV. Co-selection of antibiotic and metal resistance. Trends in Microbiology 2006;14:176–182.
17. Stokes HW, Gillings MR. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiology Reviews 2011;35:790–819.
18. Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Research 2017;27:824–834.
19. Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012;28:1420–1428.
20. Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015;31:1674–1676.
21. Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004;428:37–43.
22. Breitwieser FP, Lu J, Salzberg SL. A review of methods and databases for metagenomic classification and assembly. Briefings in Bioinformatics 2019;20:1125–1136.
23. Lu YY, Chen T, Fuhrman JA, Sun F. COCACOLA: binning metagenomic contigs using sequence COmposition, read CoverAge, CO-alignment and paired-end read LinkAge. Bioinformatics 2016;btw290.
24. Kang D, Li F, Kirton ES, Thomas A, Egan RS et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. Epub ahead of print 6 February 2019. DOI: 10.7287/peerj.preprints.27522v1.
25. Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016;32:605–607.
26. Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nature Microbiology 2018;3:836–843.
27. Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 2015;523:208–211.
28. Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nature Microbiology 2017;2:1533–1542.
29. Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R et al. The genomic and proteomic landscape of the rumen microbiome revealed by comprehensive genome-resolved metagenomics. bioRxiv. Epub ahead of print 8 December 2018. DOI: 10.1101/489443.
30. Woodcroft BJ, Singleton CM, Boyd JA, Evans PN, Emerson JB et al. Genome-centric view of carbon processing in thawing permafrost. Nature 2018;560:49–54.
31. Diamond S, Andeer PF, Li Z, Crits-Christoph A, Burstein D et al. Mediterranean grassland soil C–N compound turnover is dependent on rainfall and depth, and is mediated by genomically divergent microorganisms. Nature Microbiology 2019;4:1356–1367.
32. Meyer F, Hofmann P, Belmann P, Garrido-Oter R, Fritz A et al. AMBER: Assessment of Metagenome BinnERs. GigaScience;7. Epub ahead of print June 2018. DOI: 10.1093/gigascience/giy069.
33. Yue Y, Huang H, Qi Z, Dou H-M, Liu X-Y et al. Evaluating metagenomics tools for genome binning with real metagenomic datasets and CAMI datasets. BMC Bioinformatics;21. Epub ahead of print 28 July 2020. DOI: 10.1186/s12859-020-03667-3.
35. Soucy SM, Huang J, Gogarten JP. Horizontal gene transfer: building the web of life. Nature Reviews Genetics 2015;16:472–482.
36. Ho Sui SJ, Fedynak A, Hsiao WWL, Langille MGI, Brinkman FSL. The Association of Virulence Factors with Genomic Islands. PLoS ONE 2009;4:e8094.
37. von Wintersdorff CJH, Penders J, van Niekerk JM, Mills ND, Majumder S et al. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Frontiers in Microbiology;7. Epub ahead of print 19 February 2016. DOI: 10.3389/fmicb.2016.00173.
38. Brown-Jaque M, Calero-Cáceres W, Muniesa M. Transfer of antibiotic-resistance genes via phage-related mobile elements. Plasmid 2015;79:1–7.
39. Merkl R. SIGI: score-based identification of genomic islands. BMC Bioinformatics 2004;5:22.
40. Bertelli C, Brinkman FSL. Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics 2018;34:2161–2167.
41. Dhillon BK, Laird MR, Shay JA, Winsor GL, Lo R et al. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis: Figure 1. Nucleic Acids Research 2015;43:W104–W108.
42. Bertelli C, Tilley KE, Brinkman FSL. Microbial genomic island discovery, visualization and analysis. Briefings in Bioinformatics 2019;20:1685–1698.
43. San Millan A, Escudero JA, Gifford DR, Mazel D, MacLean RC. Multicopy plasmids potentiate the evolution of antibiotic resistance in bacteria. Nature Ecology & Evolution;1. Epub ahead of print 7 November 2016. DOI: 10.1038/s41559-016-0010.
44. San Millan A, Santos-Lopez A, Ortega-Huedo R, Bernabe-Balas C, Kennedy SP et al. Small-Plasmid-Mediated Antibiotic Resistance Is Enhanced by Increases in Plasmid Copy Number and Bacterial Fitness. Antimicrobial Agents and Chemotherapy 2015;59:3335–3341.
45. Zhou F, Xu Y. cBar: a computer program to distinguish plasmid-derived from chromosome-derived sequence fragments in metagenomics data. Bioinformatics 2010;26:2051–2052.
46. Davis JJ, Olsen GJ. Modal Codon Usage: Assessing the Typical Codon Usage of a Genome. Molecular Biology and Evolution 2009;27:800–810.
47. Daubin V, Lerat E, Perrière G.:{unav). Genome Biology 2003;4:R57.
48. Holmes AH, Moore LSP, Sundsfjord A, Steinbakk M, Regmi S et al. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 2015;387:176–87.
49. Williams KP. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Research 2002;30:866–875.
50. Schmidt H, Hensel M. Pathogenicity Islands in Bacterial Pathogenesis. Clinical Microbiology Reviews 2004;17:14–56.
51. Acuña-Amador L, Primot A, Cadieu E, Roulet A, Barloy-Hubler F. Genomic repeats, misassembly and reannotation: a case study with long-read resequencing of Porphyromonas gingivalis reference strains. BMC Genomics;19. Epub ahead of print 16 January 2018. DOI: 10.1186/s12864-017-4429-4.
52. Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S et al. Critical Assessment of Metagenome Interpretation—a benchmark of metagenomics software. Nature Methods 2017;14:1063–1071.
53. Arredondo-Alonso S, Willems RJ, van Schaik W, Schürch AC. On the (im)possibility of reconstructing plasmids from whole-genome short-read sequencing data. Microbial Genomics;3. Epub ahead of print 1 October 2017. DOI: 10.1099/mgen.0.000128.
54. Huang W, Li L, Myers JR, Marth GT. ART: a next-generation sequencing read simulator. Bioinformatics 2012;28:593–594.
55. Joshi N, Fass J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files. GitHub. https://github.com/najoshi/sickle (2011).
56. Mikheenko A, Saveliev V, Gurevich A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 2016;32:1088–1090.
57. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J et al. BLAST+: architecture and applications. BMC Bioinformatics 2009;10:421.
59. Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 2018;34:2490–2492.
60. Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009;25:1972–1973.
61. Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Molecular Biology and Evolution 2015;32:268–274.
62. Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: Combined Selection of Partitioning Schemes and Substitution Models for Phylogenetic Analyses. Molecular Biology and Evolution 2012;29:1695–1701.
63. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Research 2019;47:W256–W259.
64. Huerta-Cepas J, Serra F, Bork P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Molecular Biology and Evolution 2016;33:1635–1638.
65. Waskom M, Botvinnik O, Ostblom J, Lukauskas S, Hobson P et al.mwaskom/seaborn: v0.10.0 (January 2020). Zenodo. Epub ahead of print 24 January 2020. DOI: 10.5281/zenodo.3629446.
66. Seabold S, Perktold J. Statsmodels: Econometric and Statistical Modeling with Python. SciPy. Epub ahead of print 2010. DOI: 10.25080/majora-92bf1922-011.
67. Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics;11. Epub ahead of print 8 March 2010. DOI: 10.1186/1471-2105-11-119.
68. Alcock BP, Raphenya AR, Lau TTY, Tsang KK, Bouchard M et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Research. Epub ahead of print 29 October 2019. DOI: 10.1093/nar/gkz935.
69. Liu B, Zheng D, Jin Q, Chen L, Yang J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Research 2019;47:D687–D692.
70. Pellow D, Zorea A, Probst M, Furman O, Segal A et al. SCAPP: An algorithm for improved plasmid assembly in metagenomes. bioRxiv. Epub ahead of print 12 August 2020. DOI: 10.1101/2020.01.12.903252.
71. Giguere DJ, Bahcheli AT, Joris BR, Paulssen JM, Gieg LM et al. Complete and validated genomes from a metagenome. bioRxiv. Epub ahead of print 9 April 2020. DOI: 10.1101/2020.04.08.032540.
72. Suzuki Y, Nishijima S, Furuta Y, Yoshimura J, Suda W et al. Long-read metagenomic exploration of extrachromosomal mobile genetic elements in the human gut. Microbiome;7. Epub ahead of print 27 August 2019. DOI: 10.1186/s40168-019-0737-z.
73. Ravi A, Halstead FD, Bamford A, Casey A, Thomson NM et al. Loss of microbial diversity and pathogen domination of the gut microbiota in critically ill patients. Microbial Genomics;5. Epub ahead of print 1 September 2019. DOI: 10.1099/mgen.0.000293.
74. Liu Z, Klümper U, Liu Y, Yang Y, Wei Q et al. Metagenomic and metatranscriptomic analyses reveal activity and hosts of antibiotic resistance genes in activated sludge. Environment International 2019;129:208–220.
75. Newberry E, Bhandari R, Kemble J, Sikora E, Potnis N. Genome‐resolved metagenomics to study co‐occurrence patterns and intraspecific heterogeneity among plant pathogen metapopulations. Environmental Microbiology 2020;22:2693–2708.
76. Zhang Y, Kitajima M, Whittle AJ, Liu W-T. Benefits of Genomic Insights and CRISPR-Cas Signatures to Monitor Potential Pathogens across Drinking Water Production and Distribution Systems. Frontiers in Microbiology;8. Epub ahead of print 19 October 2017. DOI: 10.3389/fmicb.2017.02036.
77. Huang AD, Luo C, Pena-Gonzalez A, Weigand MR, Tarr CL et al. Metagenomics of Two Severe Foodborne Outbreaks Provides Diagnostic Signatures and Signs of Coinfection Not Attainable by Traditional Methods. Applied and Environmental Microbiology;83. Epub ahead of print 23 November 2016. DOI: 10.1128/aem.02577-16.
78. Klassen JL, Currie CR. Gene fragmentation in bacterial draft genomes: extent, consequences and mitigation. BMC Genomics 2012;13:14.
79. Abayasekara LM, Perera J, Chandrasekharan V, Gnanam VS, Udunuwara NA et al. Detection of bacterial pathogens from clinical specimens using conventional microbial culture and 16S metagenomics: a comparative study. BMC Infectious Diseases;17. Epub ahead of print 19 September 2017. DOI: 10.1186/s12879-017-2727-8.
80. Rogers GB, Carroll MP, Serisier DJ, Hockey PM, Jones G et al. Characterization of Bacterial Community Diversity in Cystic Fibrosis Lung Infections by Use of 16S Ribosomal DNA Terminal Restriction Fragment Length Polymorphism Profiling. Journal of Clinical Microbiology 2004;42:5176–5183.
81. Freitas AC, Chaban B, Bocking A, Rocco M, Yang S et al. The vaginal microbiome of pregnant women is less rich and diverse, with lower prevalence of Mollicutes, compared to non-pregnant women. Scientific Reports;7. Epub ahead of print 23 August 2017. DOI: 10.1038/s41598-017-07790-9.
82. Gołębiewski M, Deja-Sikora E, Cichosz M, Tretyn A, Wróbel B. 16S rDNA Pyrosequencing Analysis of Bacterial Community in Heavy Metals Polluted Soils. Microbial Ecology 2014;67:635–647.
83. Youssef N, Sheik CS, Krumholz LR, Najar FZ, Roe BA et al. Comparison of Species Richness Estimates Obtained Using Nearly Complete Fragments and Simulated Pyrosequencing-Generated Fragments in 16S rRNA Gene-Based Environmental Surveys. Applied and Environmental Microbiology 2009;75:5227–5236.
84. Claesson MJ, O’Sullivan O, Wang Q, Nikkilä J, Marchesi JR et al. Comparative Analysis of Pyrosequencing and a Phylogenetic Microarray for Exploring Microbial Community Structures in the Human Distal Intestine. PLoS ONE 2009;4:e6669.
85. Thomas M, Webb M, Ghimire S, Blair A, Olson K et al. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Scientific Reports;7. Epub ahead of print 25 September 2017. DOI: 10.1038/s41598-017-12481-6.
86. Mulvey MR, Boyd DA, Olson AB, Doublet B, Cloeckaert A. The genetics of Salmonella genomic island 1. Microbes and Infection 2006;8:1915–1922.
87. Arora SK, Wolfgang MC, Lory S, Ramphal R. Sequence Polymorphism in the Glycosylation Island and Flagellins of Pseudomonas aeruginosa. Journal of Bacteriology 2004;186:2115–2122.
88. Redondo-Salvo S, Fernández-López R, Ruiz R, Vielva L, de Toro M et al. Pathways for horizontal gene transfer in bacteria revealed by a global map of their plasmids. Nature Communications;11. Epub ahead of print 17 July 2020. DOI: 10.1038/s41467-020-17278-2.
89. Fritz A, Hofmann P, Majda S, Dahms E, Dröge J et al. CAMISIM: simulating metagenomes and microbial communities. Microbiome;7. Epub ahead of print 8 February 2019. DOI: 10.1186/s40168-019-0633-6.
90. Bokulich NA, Rideout JR, Mercurio WG, Shiffer A, Wolfe B et al. mockrobiota: a Public Resource for Microbiome Bioinformatics Benchmarking. mSystems;1. Epub ahead of print 18 October 2016. DOI: 10.1128/msystems.00062-16.
91. Karimzadeh M, Hoffman MM. Top considerations for creating bioinformatics software documentation. Briefings in Bioinformatics 2018;19:693–699.
92. Hunt M, Mather AE, Sánchez-Busó L, Page AJ, Parkhill J et al. ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microbial Genomics;3. Epub ahead of print 1 October 2017. DOI: 10.1099/mgen.0.000131.
 0000-0002-1203-9514
·
0000-0002-1203-9514
·  fmaguire
·
fmaguire
·  finlaym
finlaym